
- A-
- A+
Glossary de productos
- Cirurgia

Cirurgia
Mydriasert

Folheto informativo: Informações para o utilizador
MYDRIASERT 5.4 mg + 0.28 mg Inserto oftálmico
Cloridrato de fenilefrina + tropicamida
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico, farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
1. O que é MYDRIASERT e para que é utilizado
2. O que precisa de saber antes de utilizar MYDRIASERT
3. Como utilizar MYDRIASERT
4. Efeitos indesejáveis possíveis
5. Como conservar MYDRIASERT
6. Conteúdo da embalagem e outras informações
1. O que é MYDRIASERT e para que é utilizado
MYDRIASERT é um medicamento oftálmico, isto é, exclusivamente para tratamento ocular.
A utilização de MYDRIASERT é destinada apenas a profissionais de saúde.
MYDRIASERT destina-se a ser inserido pelo pessoal médico na pálpebra inferior do seu olho. Utiliza-se para obter uma midríase (dilatação da pupila), antes de uma intervenção cirúrgica ou para fins de diagnóstico.
2. O que precisa de saber antes de utilizar MYDRIASERT
NÃO UTILIZE MYDRIASERT nos seguintes casos:
- Se tem alergia à tropicamida ou ao cloridrato de fenilefrina ou a qualquer outro componente deste medicamente (indicados na secção 6). - Em doentes com glaucoma de ângulo fechado ou em risco de precipitação de glaucoma (aumento da pressão ocular).
- Em crianças com idade inferior a 12 anos.
Advertências e precauções
- Uma vez que este medicamento causa perturbações visuais prolongadas, tenha o cuidado de vir acompanhado à consulta (ver efeitos indesejáveis possíveis).
- Em caso de desconforto após inserção do inserto, informe o seu médico: pode ocorrer uma deslocação ou, mais raramente, a perda do inserto.
- Se sofre de grave secura dos olhos, o médico poderá ter de colocar uma gota de solução salina no olho para reduzir o risco de irritação ocular.
- Em caso de hipertensão (pressão arterial elevada), aterosclerose (espessamento da parede arterial), doença cardíaca ou hipertiroidismo (aumento da atividade da glândula tiroide) ou perturbações prostáticas, informe o seu médico.
- Em determinadas pessoas com predisposição, os agentes midriáticos (produtos que dilatam a pupila) podem desencadear uma crise de glaucoma agudo (devido ao súbito aumento da pressão ocular).
- É desaconselhado o uso de lentes de contacto hidrófilas durante o tratamento.
Crianças e adolescentes
Mydriasert não deve ser utilizado em crianças com idade inferior a 12 anos, uma vez que as crianças parecem ser mais sensíveis ao risco de efeitos indesejáveis graves. Não se recomenda a utilização de Mydriasert em crianças com idades entre os 12 e os 18 anos pois não existe experiência clínica adequada.
Outros medicamentos e MYDRIASERT
Informe o seu médico se estiver a usar medicamentos que dilatem as pupilas (midríase), para além de Mydriasert, de forma que o médico possa determinar a quantidade total de agente midriático à qual ficará exposto.
Informe o seu médico ou farmacêutico se estiver a tomar, tiver tomado recentemente ou se vier a tomar outros medicamentos, incluindo medicamentos obtidos sem receita médica. Embora Mydriasert seja aplicado localmente, estas informações são importantes, uma vez que alguns medicamentos podem afetar a ação de outros.
Gravidez e aleitamento
Não existem dados adequados sobre a utilização de Mydriasert ou das suas substâncias ativas em mulheres grávidas. Por isso, Mydriasert não deverá ser utilizado durante a gravidez, a menos que seja claramente necessário.
A utilização durante o aleitamento não é recomendada.
Condução de veículos e utilização de máquinas
Não conduza ou utilize máquinas porque MYDRIASERT pode causar perturbações visuais incómodas por algumas horas (turvação devido à dilatação prolongada da pupila).
Informações importantes sobre alguns componentes de MYDRIASERT
Os desportistas devem ser alertados para o facto de este medicamento conter uma substância ativa (o cloridrato de fenilefrina) que pode originar resultados positivos nos testes para deteção de substâncias proibidas.
3. Como utilizar MYDRIASERT
DESTINADO APENAS A UTILIZAÇÃO POR PROFISSIONAIS DE SAÚDE.
Mydriasert destina-se a ser usado em adultos. Mydriasert não deve ser ingerido.
O profissional de saúde colocará um inserto oftálmico dentro da pálpebra inferior do olho em questão. O profissional retirará o inserto em 30 minutos, quando a pupila se apresentar suficientemente dilatada, e antes de proceder à operação ou à investigação do olho. O inserto oftálmico não deve permanecer dentro do olho por mais de 2 horas.
Se utilizar mais MYDRIASERT do que deveria
Dado que um único inserto será inserido no olho pelo médico ou profissional de saúde, é improvável o risco de utilizar mais Mydriasert do que o recomendado. No entanto, se o profissional de saúde precisar de usar gotas oculares midriáticas, para além de Mydriasert, pode ocorrer o risco de sobredosagem das substâncias ativas de Mydriasert.
Os sintomas de sobredosagem das substâncias ativas de Mydriasert podem incluir fadiga extrema, sudação, tonturas, diminuição do ritmo cardíaco, coma, dores de cabeça, aumento do ritmo cardíaco, boca e pele seca, sonolência invulgar, rubor e dilatação prolongada das pupilas.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Frequentes: podem afetar até 1 em 10 pessoas:
- picadas,
- visão enevoada,
- desconforto visual devido à perceção da presença ou deslocação do inserto oftálmico.
Pouco frequentes: podem afetar até 1 em 100 pessoas:
- lacrimejo,
- irritação,
- encadeamento devido à dilatação prolongada da pupila,
- queratite ponteada superficial (inflamação da córnea).
Raros: podem afetar até 1 em 1.000 pessoas:
- reações alérgicas: inflamação das pálpebras (blefarite), inflamação da conjuntiva (conjuntivite).
Muito raros: podem afetar até 1 em 10.000 pessoas:
- convulsões.
Foram observadas úlceras da córnea (pequenas erosões da superfície do olho) e edema da córnea (inflamação da superfície do olho) quando o inserto oftálmico foi acidentalmente deixado dentro do olho.
Em indivíduos com predisposição, MYDRIASERT pode desencadear uma crise de glaucoma agudo (súbito aumento da pressão intraocular):em caso de sintomas anormais após a administração (vermelhidão, dor e perturbações visuais), contacte imediatamente o seu médico.
Embora improvável após administração no olho, as substâncias ativas contidas em MYDRIASERT podem causar os seguintes efeitos secundários, que deverão ser tidos em consideração:
- aumento da pressão arterial, taquicardia,
- muito raramente, incidentes mais graves, como arritmia cardíaca,
- tremores, palidez, dores de cabeça, boca seca.
Efeitos indesejáveis adicionais em crianças
Frequência desconhecida (não pode ser calculada a partir dos dados disponíveis)
- Fluido ou inchaço nos pulmões
- Palidez em redor dos olhos em bebés prematuros
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar MYDRIASERT
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na saqueta após "VAL".
Não conservar acima de 25°C.
Usar imediatamente após abertura da saqueta.
Não utilizar MYDRIASERT se observar qualquer deterioração no invólucro da saqueta ou no inserto oftálmico.
6. Conteúdo da embalagem e outras informações
Qual a composição de MYDRIASERT
- As substâncias ativas são tropicamida 0.28 mg e cloridrato de fenilefrina 5.4 mg, em cada inserto oftálmico.
- Os outros componentes são o metacrilato de amónio copolímero tipo A, Eudragit NE 30 D, dibehenato de glicerilo e etilcelulose.
Qual o aspeto de MYDRIASERT e conteúdo da embalagem
MYDRIASERT é fornecido numa saqueta.
MYDRIASERT tem o formato de uma pequena placa (inserto) branca, oblonga (4.3 mm x 2.3 mm).
Cada embalagem contém outra saqueta com uma pinça esterilizada descartável, usada para colocação de MYDRIASERT no olho.
Embalagens de 1 inserto e 1 pinça, 10 insertos e 10 pinças, 20 insertos e 20 pinças, 50 insertos e 50 pinças ou 100 insertos e 100 pinças.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires Théa
12, Rue Louis Blériot - Zone Industrielle du Brézet
63017 CLERMONT-FERRAND – FRANÇA
Fabricante:
BENAC
10, Avenue Paul Langevin
17180 PÉRIGNY – FRANÇA
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
| Alemanha | Mydriasert |
| Áustria | Mydriasert |
| Bélgica | Mydriasert |
| Dinamarca | Mydriasert |
| Espanha | Mydriasert |
| Finlândia | Mydriasert |
| França | Mydriasert |
| Holanda | Mydriasert |
| Itália | Mydriasert |
| Luxemburgo | Mydriasert |
| Portugal | Mydriasert |
|
Reino Unido
|
Mydriasert |
| Suécia | Mydriasert |
Este folheto informativo foi revisto pela última vez em 11/2021
_______________________________________________________________
A informação que se segue destina-se apenas aos médicos e aos profissionais dos cuidados de saúde.
Não ingerir.
Antes de usar o medicamento, verifique a integridade da saqueta. Em caso de deterioração do invólucro da saqueta, a esterilidade deixa de estar assegurada; neste caso, use outro inserto oftálmico retirado de uma embalagem intacta.
POSOLOGIA
O profissional de saúde coloca um inserto oftálmico no saco conjuntival inferior do olho em questão, num máximo de duas horas antes da intervenção cirúrgica ou da investigação.
População pediátrica
Mydriasert está contraindicado em crianças com idade inferior a 12 anos.
Não existem dados quanto à utilização em crianças com idades entre os 12 e os 18 anos, pelo que Mydriasert não é recomendado para estes doentes.
MODO DE ADMINISTRAÇÃO
Cortar o bordo do invólucro ao longo da linha ponteada, abrir a saqueta e localizar o inserto. Puxar para baixo a pálpebra inferior, segurando-a entre o polegar e o indicador (A) e aplicar o inserto oftálmico no saco conjuntival inferior, usando a pinça esterilizada descartável fornecida na embalagem, a qual deve ser eliminada imediatamente após a utilização (B).
INSTRUÇÕES DE UTILIZAÇÃO
O inserto oftálmico não deve ser deixado mais de duas horas dentro do saco conjuntival inferior. O médico pode retirar o inserto oftálmico logo que a midríase seja considerada suficiente para a intervenção ou o procedimento a realizar; no máximo, o inserto deve ser retirado 30 minutos após dilatação suficiente da pupila. Em caso de desconforto, verificar se o inserto foi corretamente inserido na base do saco conjuntival inferior.
ADVERTÊNCIA: RETIRADA DO INSERTO OFTÁLMICO
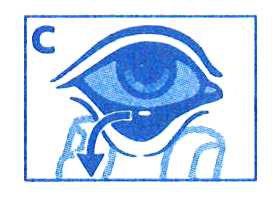
Antes de uma intervenção ou procedimento, e logo que se obtenha a midríase desejada, o inserto oftálmico deve ser retirado do saco conjuntival inferior usando uma pinça cirúrgica esterilizada ou uma mecha esterilizada ou uma solução de lavagem ou irrigação esterilizada, puxando para baixo a pálpebra inferior (C).


Cirurgia
Aprokam

Folheto informativo: Informação para o utilizador
APROKAM 50 mg pó para solução injetável
Cefuroxima
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
1. O que é APROKAM e para que é utilizado
2. O que precisa de saber antes de lhe ser administrado APROKAM
3. Como é administrado APROKAM
4. Efeitos secundários possíveis
5. Como conservar APROKAM
6. Conteúdo da embalagem e outras informações
1. O que é APROKAM e para que é utilizado
• APROKAM contém uma substância ativa, cefuroxima (sob a forma de cefuroxima sódica), que pertence a um grupo de antibióticos chamados cefalosporinas. Os antibióticos são usados para matar as bactérias ou “germes” que causam infeções.
• Este medicamento será utilizado se for submetido a uma cirurgia aos olhos por ter cataratas (enevoamento do cristalino).
• O seu cirurgião oftalmológico administrará este medicamento através de uma injeção no olho no final da cirurgia da catarata para prevenir uma infeção no olho.
2. O que precisa de saber antes de lhe ser administrado APROKAM
Não utilize APROKAM:
- se tem alergia (hipersensibilidade) à cefuroxima ou a qualquer outro antibiótico do tipo das cefalosporinas.
Advertências e precauções
Fale com o seu médico ou farmacêutico ou enfermeiro antes de utilizar APROKAM:
• se é alérgico a outros antibióticos como a penicilina,
• se está em risco de contrair uma infeção devido a uma bactéria chamada Staphylococcus aureus resistente à meticilina,
• se está em risco de contrair uma infeção grave,
• se lhe foi diagnosticada uma catarata complicada,
• se está planeada uma cirurgia ocular combinada,
• se tem uma doença grave da tiroide.
APROKAM é apenas administrado através de uma injeção no olho (injeção intracameral).
APROKAM deve ser administrado nas condições de assepsia (o que significa em condições de higiene e isentas de germes) da cirurgia da catarata.
Um frasco para injetáveis de APROKAM deve ser usado apenas para um doente.
Outros medicamentos e APROKAM
Informe o seu médico ou farmacêutico se estiver a tomar, tiver tomado recentemente ou se vier a tomar outros medicamentos.
Gravidez e amamentação
• Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de lhe ser administrado este medicamento.
• APROKAM ser-lhe-á apenas administrado se os benefícios compensarem os possíveis riscos.
APROKAM contém sódio
Este medicamento contém menos do que 1 mmol (23 mg) de sódio por dose ou seja, é praticamente “isento de sódio”.
3. Como é administrado APROKAM
• As injeções de APROKAM serão administradas por um cirurgião oftalmológico no final da cirurgia da catarata.
• APROKAM é fornecido sob a forma de um pó estéril que, antes de ser administrado, é dissolvido numa solução de soro fisiológico para injetáveis.
Se lhe for administrado menos ou mais APROKAM do que deveria
A sua medicação ser-lhe-á geralmente administrada por um profissional de saúde. Se pensa que se podem ter esquecido de lhe administrar uma dose ou que recebeu uma dose do medicamento superior à que deveria, por favor informe o seu médico ou enfermeiro.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico ou enfermeiro.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
O seguinte efeito secundário é muito raro (pode afetar até 1 em 10.000 pessoas):
• Reação alérgica grave que provoca dificuldade em respirar ou tonturas.
O seguinte efeito secundário é reportado com uma frequência “Desconhecida” (não pode ser calculada a partir dos dados disponíveis):
• Edema macular (visão turva ou distorcida na proximidade ou no centro do seu campo de visão).
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar APROKAM
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize APROKAM após o prazo de validade impresso no rótulo do frasco para injetáveis e na embalagem exterior após EXP. O prazo de validade corresponde ao último dia do mês indicado.
Conservar abaixo de 25ºC. Manter o frasco para injetáveis na embalagem exterior para proteger da luz.
Apenas para uma única utilização.
Após reconstituição: o produto deve ser usado imediatamente.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de APROKAM
- A substância ativa é a cefuroxima (sob a forma de cefuroxima sódica).
- Cada frasco para injetáveis contém 50 mg de cefuroxima.
- Após reconstituição, 0,1 ml de solução contêm 1 mg de cefuroxima.
- Não contém outros componentes.
Qual o aspeto de APROKAM e conteúdo da embalagem
APROKAM é um pó para solução injetável branco ou esbranquiçado, fornecido num frasco para injetáveis de vidro.
Cada embalagem contém um ou dez ou vinte frascos para injetáveis ou 10 frascos para injetáveis com 10 agulhas estéreis com filtro. É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
LABORATOIRES THEA
12 Rue Louis Blériot
63017 CLERMONT-FERRAND Cedex 2
França
Fabricante
BIOPHARMA S.R.L.
Via Delle Gerbere, 22/30 (loc. S. PALOMBA)
00134 ROMA (RM)
Itália
ou
LABORATOIRES THEA
12 Rue Louis Blériot
63017 CLERMONT-FERRAND Cedex 2
França
Este medicamento encontra-se autorizado nos seguintes Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Alemanha, Áustria, Bélgica, Bulgária, Dinamarca, Eslováquia, Eslovénia, Finlândia, França, Holanda, Itália, Islândia, Luxemburgo, Noruega, Polónia, Portugal, República Checa, Roménia, Suécia, Reino Unido: APROKAM
Chipre, Grécia, Espanha: PROKAM
Irlanda: APROK
Este folheto foi revisto pela última vez em 08/2019
A informação que se segue destina-se apenas aos profissionais de saúde:
Incompatibilidades
Não se encontra descrita na literatura qualquer incompatibilidade com os produtos mais frequentemente utilizados na cirurgia da catarata. Este medicamento não deve ser misturado com outros medicamentos, exceto os que se encontram mencionados em seguida [solução injetável de cloreto de sódio a 9 mg/ml (0,9%)].
Como preparar e administrar APROKAM
Frasco para injetáveis para uma única utilização apenas para administração intracameral.
APROKAM deve ser administrado após reconstituição através de uma injeção intraocular na câmara anterior do olho (injeção intracameral) efetuada por um cirurgião oftalmológico, nas condições de assepsia recomendadas para a cirurgia da catarata.
A solução reconstituída deve ser visualmente inspecionada e só deverá ser utilizada se a solução for incolor ou ligeiramente amarelada e isenta de partículas visíveis.
O produto deve ser usado imediatamente após a reconstituição e não deve ser reutilizado.
A dose recomendada para a cefuroxima é de 1 mg em 0,1 ml de solução injetável de cloreto de sódio a 9 mg/ml (0,9%).
NÃO INJETE UMA DOSE SUPERIOR À RECOMENDADA.
O frasco para injetáveis destina-se apenas a uma única utilização.
Um frasco para injetáveis apenas para um doente. Cole a etiqueta destacável do frasco para injetáveis na ficha do doente.
Siga as seguintes instruções para preparar o APROKAM para administração intracameral:
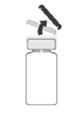
1. Verifique a integridade da cápsula de abertura fácil antes de a retirar.
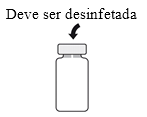
2. Desinfete a superfície da tampa de borracha antes de efetuar o passo 3.
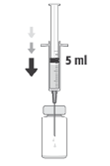
3. Insira a agulha na vertical no centro da tampa do frasco para injetáveis, mantendo o frasco para injetáveis na posição vertical. Em seguida, usando uma técnica asséptica injete no frasco para injetáveis 5 ml de uma solução injetável de cloreto de sódio a 9 mg/ml (0,9%).
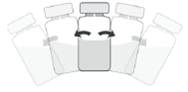
4. Agite suavemente até que a solução esteja isenta de partículas visíveis.
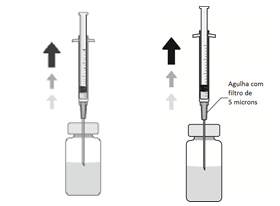
5. Adapte uma agulha estéril (18G x 1½”, 1,2 mm x 40 mm) com um filtro de 5 microns (membrana de copolímero acrílico num suporte de nylon não tecido) numa seringa estéril de 1 ml (a agulha estéril com um filtro de 5 microns poderá ser fornecida na embalagem). Empurre esta seringa verticalmente no centro da tampa do frasco para injetáveis, mantendo o frasco para injetáveis na posição vertical.
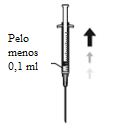
6. Aspire assepticamente, pelo menos 0,1 ml de solução.
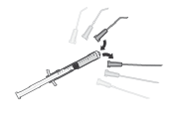
7. Retire a agulha da seringa e adapte na seringa uma cânula apropriada para a câmara anterior.

8. Cuidadosamente deve expelir o ar da seringa e ajustar a dose para atingir a marca correspondente a 0,1 ml na seringa. A seringa encontra-se preparada para a injeção.
Após a utilização, rejeite a solução reconstituída que ainda restar. Não a guarde para uma utilização subsequente.
Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com as exigências locais. Elimine as agulhas utilizadas nos contentores adequados.

Cirurgia
Mydrane

Folheto informativo: Informação para o doente
MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml solução injetável
Tropicamida / cloridrato de fenilefrina / cloridrato de lidocaína mono-hidratado
Leia com atenção todo este folheto antes de lhe ser administrado este medicamento, pois contém informação importante para si.
Conserve este folheto. Pode ter necessidade de o ler novamente.
Caso ainda tenha dúvidas, fale com o seu médico, farmacêutico ou enfermeiro.
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
1. O que é MYDRANE e para que é utilizado
2. O que precisa de saber antes de lhe ser administrado MYDRANE
3. Como é administrado MYDRANE
4. Efeitos secundários possíveis
5. Como conservar MYDRANE
6. Conteúdo da embalagem e outras informações
1. O que é MYDRANE e para que é utilizado
O que é MYDRANE
Este medicamento é uma solução, a qual lhe vai ser injetada no olho.
Contém 3 substâncias ativas:
• tropicamida que pertence a um grupo de medicamentos que bloqueia a passagem de impulsos por determinados nervos (conhecido como anticolinérgico),
• fenilefrina (sob a forma de cloridrato de fenilefrina) que pertence a um grupo de medicamentos que mimetiza os efeitos dos impulsos transmitidos por determinados nervos (o qual pertence aos simpaticomiméticos alfa),
• lidocaína (sob a forma de cloridrato de lidocaína mono-hidratado) que pertence a uma classe de medicamentos denominada anestésicos locais do tipo amida.
Para que é utilizado
Este medicamento é utilizado apenas em adultos.
Ser-lhe-á administrado pelo seu cirurgião oftalmológico através de uma injeção no seu olho no início da cirurgia da catarata (enevoamento do cristalino), para dilatar a pupila do seu olho (midríase) e anestesiar o seu olho durante o procedimento cirúrgico.
2. O que precisa de saber antes de lhe ser administrado MYDRANE
Não lhe deve ser administrado MYDRANE:
• se tem alergia à tropicamida, ao cloridrato de fenilefrina e/ou ao cloridrato de lidocaína mono-hidratado ou a qualquer outro componente deste medicamento (indicados na secção 6),
• se tem alergia aos anestésicos do tipo amida (articaína, bupivacaína, mepivacaína, prilocaína, ropivacaína),
• se tem alergia aos derivados da atropina.
Advertências e precauções
MYDRANE não é recomendado:
• na cirurgia da catarata quando combinada com um certo tipo de cirurgia ocular (vitrectomia),
• se a parte anterior (câmara anterior) do seu olho está inchada,
• se tem antecedentes de pressão ocular elevada aguda (glaucoma de angulo estreito agudo).
Deve falar com o seu médico especialmente se tiver:
• pressão arterial elevada (hipertensão),
• espessamento da parede arterial (aterosclerose),
• qualquer doença no coração e em particular se esta afetar o ritmo cardíaco,
• uma contraindicação para medicamentos que aumentem a pressão arterial (aminas vasopressoras: adrenalina, noradrenalina, dopamina, dobutamina) por via oral,
• hiperatividade da glândula tiroide (hipertiroidismo),
• perturbações na glândula da próstata,
• crises de epilepsia,
• qualquer doença no fígado ou problemas nos rins,
• qualquer problema com a sua respiração,
• perda da função muscular e fraqueza (miastenia gravis).
Outros medicamentos e MYDRANE
Informe o seu médico ou farmacêutico se estiver a tomar, tiver tomado recentemente ou se vier a tomar outros medicamentos.
Gravidez, amamentação e fertilidade
Este medicamento não deve ser utilizado:
• durante a gravidez,
• durante a amamentação.
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento.
Condução de veículos e utilização de máquinas
MYDRANE tem uma influência moderada na capacidade de conduzir e utilizar máquinas. Consequentemente, não deve conduzir e/ou utilizar máquinas até que a sua visão volte ao normal.
MYDRANE contém sódio
Este medicamento contém menos do que 1 mmol (23 mg) de sódio por unidade de dose, ou seja, é praticamente “isento de sódio”.
3. Como é administrado MYDRANE
Apenas lhe será administrado este medicamento caso já tenha demonstrado, numa avaliação pré-operatória, uma dilatação satisfatória da pupila com uma terapêutica midriática tópica.
Dose e modo de administração
• MYDRANE injetável ser-lhe-á administrado por um cirurgião oftalmológico, sob anestesia local, no início da cirurgia da catarata.
• A dose recomendada é de 0,2 ml de solução, numa injeção única. Não deverá ser administrada uma dose adicional, pois não foi demonstrado efeito adicional e além disso foi observado um aumento da perda de células endoteliais (células da camada que reveste a superfície posterior da córnea).
• É utilizada a mesma dose para adultos e idosos.
Se lhe for administrado mais, ou menos, MYDRANE do que deveria
O seu medicamento ser-lhe-á administrado por um cirurgião oftalmológico. É pouco provável que lhe seja administrada uma dose excessiva.
A sobredosagem pode causar um aumento na perda de células endoteliais (células da camada que cobre a superfície posterior da córnea).
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico ou enfermeiro.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Complicações mais graves e bem conhecidas que ocorrem durante ou após a cirurgia da catarata:
Pouco frequentes: podem afetar até 1 em 100 pessoas
• Lesão do cristalino (rutura da cápsula posterior),
• Inchaço da retina (edema macular cistoide).
Nesta situação procure aconselhamento médico urgente.
Outros efeitos secundários:
Pouco frequentes: podem afetar até 1 em 100 pessoas
• Dores de cabeça,
• Inchaço da córnea (queratite), pressão no olho aumentada, vermelhidão do olho (hiperemia ocular),
• Pressão arterial elevada (hipertensão).
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar MYDRANE
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na cartonagem, blister e ampola. O prazo de validade corresponde ao último dia do mês indicado.
O medicamento não necessita de quaisquer precauções especiais de conservação.
Apenas para uma utilização única. Este medicamento deve ser utilizado imediatamente após a primeira abertura da ampola.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de MYDRANE
• As substâncias ativas são: tropicamida 0,04 mg, cloridrato de fenilefrina 0,62 mg e cloridrato de lidocaína mono-hidratado 2 mg por cada dose de 0,2 ml, o que corresponde a 0,2 mg de tropicamida, 3,1 mg de cloridrato de fenilefrina e 10 mg de cloridrato de lidocaína mono-hidratado por cada 1 ml.
• Os outros componentes são: cloreto de sódio, fosfato disódico dodeca-hidratado, fosfato disódico di-hidratado, edetato dissódico, água para preparações injetáveis.
Qual o aspeto de MYDRANE e conteúdo da embalagem
MYDRANE é uma solução injetável límpida, ligeiramente amarela-acastanhada, praticamente isenta de partículas visíveis, acondicionada numa ampola de vidro castanho de 1 ml. Cada ampola estéril contém 0,6 ml de solução injetável e encontra-se acondicionada separadamente ou em conjunto com uma agulha estéril com filtro de 5 micrómetros num blister selado papel/PVC.
Cada embalagem contém 1 ou 20 ou 100 ampolas estéreis com agulha(s) estéril(eis) com filtro de 5 micrómetros fornecidas separadamente ou em conjunto no mesmo blister. As agulhas com filtro de 5 micrómetros devem ser utilizadas apenas para retirar o conteúdo da ampola. Todos os componentes são apenas para uma única utilização.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires THEA
12, Rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
Fabricante
Delpharm Tours
Rue Paul Langevin
37170 Chambray les tours
França
ou
Laboratoires Thea
12, Rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
Este medicamento encontra-se autorizado nos seguintes Estados membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Alemanha, Áustria, Bélgica, Bulgária, Chipre, Croácia, Dinamarca, Eslováquia, Eslovénia, Finlândia, França, Grécia, Holanda, Itália, Irlanda, Luxemburgo, Polónia, Portugal, Roménia, Reino Unido, República Checa, Suécia Mydrane
Espanha, Irlanda Fydrane
Noruega Mydane
Este folheto foi revisto pela última vez em 09/2023
A informação que se segue destina-se apenas aos profissionais de saúde:
Incompatibilidades
Não foram reportadas na literatura nem durante os ensaios clínicos, incompatibilidades entre os produtos mais frequentemente utilizados na cirurgia da catarata e as substâncias ativas. Para os viscoelásticos comuns, tal foi também confirmado pelos de testes de interação farmacêutica.
Advertência
Não utilizar se o blister estiver danificado ou rasgado. Abrir apenas sob condições asséticas. Garante-se que o conteúdo do blister se encontra estéril.
Como preparar e administrar MYDRANE
Solução para utilização apenas num olho por via intracameral.
MYDRANE deve ser administrado por injeção intraocular na câmara anterior do olho (injeção intracameral), por um cirurgião oftalmológico, nas condições de assepsia recomendadas para a cirurgia da catarata.
Antes da injeção intracameral, a solução deve ser visualmente inspecionada e só deverá ser utilizada se estiver límpida ligeiramente amarela-acastanhada e praticamente isenta de partículas visíveis.
A dose recomendada é de 0,2 ml de MYDRANE; não deverá ser administrada uma dose adicional, pois não foi demonstrado efeito adicional e foi observado um aumento da perda de células endoteliais.
O produto deve ser utilizado imediatamente após a primeira abertura da ampola e não deve ser reutilizado em outro olho ou em qualquer outro doente.
Apenas para a apresentação em kit (i.e. blister contendo uma ampola e uma agulha): cole a etiqueta do blister no processo do doente.
Siga as seguintes instruções para preparar MYDRANE para a injeção intracameral:
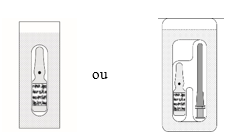
1. Verifique o blister antes de o abrir para garantir que está intacto. Retire a película do blister em condições asséticas para garantir a esterilidade do conteúdo.

2. Abra a ampola estéril que contém o produto. A ampola deve ser aberta pelo ponto de quebra (OPC) da seguinte forma: segure a parte de baixo da ampola com o polegar a apontar para o ponto colorido. Segure o topo da ampola com a outra mão, posicionando o polegar no ponto colorido e pressione para trás de forma a quebrar no ponto de quebra que está abaixo do ponto colorido.
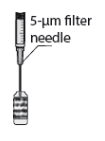
3. Encaixe a agulha estéril com filtro de 5 microns (fornecida) numa seringa estéril. Retire a proteção da agulha estéril com filtro de 5 microns e aspire, pelo menos, 0,2 ml de solução injetável da ampola para a seringa.
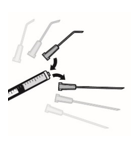
4. Retire a agulha da seringa e adapte na seringa uma cânula apropriada para a câmara anterior.
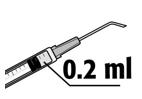
5. Cuidadosamente deve expelir o ar da seringa. Ajuste até aos 0,2 ml. A seringa encontra-se pronta para ser utilizada.
6. Injete lentamente os 0,2 ml contidos na seringa na câmara anterior do olho, numa injeção única, através da paracentese ou da incisão principal.
Após a utilização, rejeite a solução que ainda reste. Não a guarde para uma utilização subsequente.
Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com as exigências locais. Elimine as agulhas utilizadas nos contentores adequados.