
- A-
- A+
Produtos
Os nossos produtos em Portugal.
- Alergia

Alergia
Cromabak

Folheto informativo: Informação para o utilizador
Cromabak 20mg/ml,
Colírio, solução
Cromoglicato de sódio
Leia com atenção todo estefolheto antes de começar a utilizareste medicamento pois contém informação importante para si.
- Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico ou farmacêutico.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso precise de esclarecimentos ou conselhos, consulte o seu farmacêutico.
- Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico ou farmacêutico. Ver secção 4.
- Se não se sentir melhor ou se piorar, tem de consultar um médico.
O que contém este folheto:
- O que é CROMABAK e para que é utilizado
- O que precisa de saber antes de utilizar CROMABAK
- Como utilizar CROMABAK
- Efeitos indesejáveis possíveis
- Como conservar CROMABAK
- Conteúdo da embalagem e outras informações
1. O que é CROMABAK e para que é utilizado
Este colírio está indicado no tratamento da prevenção dos sintomas de conjuntivite alérgica perene ou sazonal com diagnóstico médico prévio.
Se não se sentir melhor ou se piorar, tem de consultar um médico.
2. O que precisa de saber antes deutilizar CROMABAK
Não utilize CROMABAK
- Se tem alergia ao cromoglicato de sódio ou a qualquer outro componente deste medicamento (indicados na secção 6).
- Sem consultar o seu médico se tiver doença renal ou hepática grave.
Advertências e precauções
Fale com o seu médico ou farmacêutico antes de utilizar CROMABAK.
- Não injete e não ingira.
- Lave cuidadosamente as mãos antes de efetuar a instilação.
- Evite o contacto da extremidade do frasco com o olho ou com as pálpebras.
- Volte a colocar a tampa do frasco após a utilização.
- Não ultrapasse a posologia recomendada.
- Depois de aberto o frasco não deve utilizar o colírio por um período superior a oito semanas.
- Em caso de tratamento simultâneo com outro colírio, espere 15 minutos entre as instilações dos dois colírios.
- A ausência de conservante permite a utilização do colírio pelos portadores de lentes de contacto.
Outros medicamentos e CROMABAK
Informe o seu médico ou farmacêutico se estiver a utilizar, ou tiver utilizado recentemente, ou se vier a utilizar outros medicamentos.
Em caso de tratamento simultâneo com outro colírio, espere 15 minutos entre as instilações dos dois colírios.
Se estiver a ser medicado com outros medicamentos, pergunte ao seu médico se pode utilizar este.
Gravidez e amamentação
Consulte o seu médico ou farmacêutico antes de tomar qualquer medicamento.
Não há estudos da utilização de CROMABAK colírio por mães a amamentar pelo que durante este período só o médico pode fazer com precaução a avaliação do benefício-risco.
Se está grávida ou amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento.
Condução de veículos e utilização de máquinas
Tal como acontece com todos os colírios, a sua instilação pode causar visão turva.
Cromabak contém fosfatos
Este medicamento contém 0,0160 mg de fosfatos em cada gota, que é equivalente a 0,5193 mg/ml (relativamente aos seus efeitos, ver secção 4).
3. Como utilizar CROMABAK
Utilize este medicamento exatamente como indicado pelo seu médico ou farmacêutico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
A dose recomendada é:
Adultos e crianças com idade superior a 4 anos: em função da gravidade dos sintomas, instilar 1 gota no olho afetado, 4 a 6 vezes por dia, com intervalos regulares.
- Lavar cuidadosamente as mãos antes de efetuar a instilação.
- Instilar uma gota de colírio no saco conjuntival inferior, puxando a pálpebra ligeiramente para baixo e olhando para cima.
- Voltar a colocar a tampa do frasco após a utilização.
Este medicamento não é para uso prolongado. Se até 7 dias os sintomas não passarem, deve contactar o seu médico.
Fale com o seu médico ou farmacêutico se tiver a impressão de que CROMABAK é demasiado forte ou demasiado fraco.
Se utilizar mais CROMABAK do que deveria
Consulte imediatamente o seu médico ouo seu farmacêutico.
Caso se tenha esquecido de utilizar CROMABAK
Não instile uma dose a dobrar para compensar a dose que se esqueceu de instilar e continue o seu tratamento conforme recomendado.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos indesejáveis possíveis
Como todos os medicamentos, este medicamento pode causar efeitos indesejáveis, embora estes não se manifestem em todas as pessoas. Tal é o caso da possível ocorrência de reações alérgicas ao cromoglicato de sódio ou a qualquer outro componente do medicamento.
Com a instilação do colírio pode ocorrer, desconforto visual transitório (sensação de queimadura e picadas nos olhos).
Cromabak contém fosfatos. Se sofrer de lesão grave na camada transparente na parte da frente do olho (córnea), os fosfatos podem causar em casos muito raros zonas nubladas na córnea devido à acumulação de cálcio durante o tratamento.
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico ou farmacêutico. Também poderá comunicar efeitos indesejáveis diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos indesejáveis, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
(preferencialmente)
ou através dos seguintes contactos:
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar CROMABAK
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior e no frasco, após VAL. O prazo de validade corresponde ao último dia do mês indicado.
Conservar a temperatura inferior a 25ºC.
Manter o frasco dentro da embalagem exterior para proteger da luz.
Qualquer frasco uma vez aberto, não deve ser conservado para além de 8 semanas após a primeira abertura.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como eliminar os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de CROMABAK?
- A substância ativa é o cromoglicato de sódio a 20 mg/ml.
- Os outros componentes são: sorbitol, fosfato dissódico dodeca-hidratado, fosfato monossódicodi-hidratado e água para preparações injetáveis.
Qual o aspeto de CROMABAK e conteúdo da embalagem?
CROMABAK apresenta-se na forma de uma solução incolor ou ligeiramente amarelada, em frascos conta-gotas de polietileno, contendo 5 ou 10 ml de colírio.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires Théa
12, Rue Louis Blériot
Zone Industrielle du Brézet
63017 Clermont-Ferrand
França
Fabricantes
Excelvision
Rue de la Lombardière
07100 Annonay
França
Farmila - TheaFarmaceuticiS.p.A.
Via Enrico Fermi, 50 SettimoMilanese - Milano
Itália
Este folheto foi revisto pela última vez em 12/2022

Alergia
Naabak

Folheto informativo: Informação para o utilizador
NAABAK 49 mg/ml, colírio, solução
Ácido N-acetil-aspartil-glutâmico
Leia com atenção todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, informe o seu médico ou farmacêutico. Ver secção 4.
O que contém este folheto:
1. O que é NAABAK e para que é utilizado
2. O que precisa de saber antes de utilizar NAABAK
3. Como utilizar NAABAK
4. Efeitos secundários possíveis
5. Como conservar NAABAK
6. Conteúdo da embalagem e outras informações
1. O que é NAABAK e para que é utilizado
Por aplicação ocular, o ácido N-aceti1-aspartil-glutâmico exerce um efeito local, e previne assim as reações inflamatórias alérgicas oculares, desencadeadas pelos alérgenos (pólen, pó das casas, etc..).
Este medicamento está indicado para alívio dos sintomas oculares de origem alérgica (conjuntivites e blefaroconjuntivites).
2. O que precisa de saber antes de utilizar NAABAK
Não utilize NAABAK
- se tem alergia ao ácido N-aceti1-aspartil-glutâmico ou a qualquer outro componente deste medicamento (indicados na secção 6).
Advertências e precauções
- Fale com o seu médico ou farmacêutico antes de utilizar NAABAK.
- Não injetar e não ingerir.
- Lavar cuidadosamente as mãos antes de efetuar a instilação.
- Evitar o contacto da extremidade do frasco com o olho ou com as pálpebras.
- A inexistência de conservante nas gotas instiladas permite a utilização do colírio pelos portadores de lentes de contacto.
- Em crianças com menos de 4 anos de idade, não utilizar sem recomendação médica.
- Em caso de alergia conjuntival grave, consultar o seu médico.
- Não ultrapassar a posologia prescrita e não interromper o tratamento sem recomendação médica.
Outros medicamentos e NAABAK
Em caso de tratamento simultâneo com outro colírio, esperar 15 minutos entre as instilações dos dois colírios.
Informe o seu médico ou farmacêutico se estiver a tomar, ou tiver tomado recentemente, ou se vier a tomar outros medicamentos.
Gravidez e amamentação
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de utilizar este medicamento.
Não é recomendado o uso deste medicamento durante a gravidez.
Não é recomendado o uso deste medicamento durante o período de aleitamento.
Condução de veículos e utilização de máquinas
Os doentes que apresentem visão enevoada não devem conduzir ou utilizar máquinas.
3. Como utilizar NAABAK
Utilize este medicamento exatamente como indicado pelo seu médico. Fale com o seu médico ou farmacêutico se tiver dúvidas. A dose recomendada é de 1 gota, 2 a 6 vezes por dia.
Lavar cuidadosamente as mãos antes da instilação.
Evitar o contacto entre a extremidade do conta-gotas e o olho ou as pálpebras.
Instilar a gota de colírio no saco conjuntival inferior, puxando ligeiramente para baixo a pálpebra inferior e olhando para cima.
O tempo de aparecimento de cada gota é mais longo do que num frasco de colírio clássico.
Colocar a tampa no frasco após a utilização.
Caso não haja recomendação médica diferente, o tratamento deve terminar logo que desapareçam os sintomas de alergia.
Fale com o seu médico ou farmacêutico se tiver a impressão de que NAABAK é demasiado forte ou demasiado fraco.
Se utilizar mais NAABAK do que deveria
Consulte imediatamente o seu médico ou o seu farmacêutico.
Caso se tenha esquecido de utilizar NAABAK
Não instile uma dose a dobrar para compensar uma dose que se esqueceu de instilar e continue o seu tratamento conforme recomendado.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Breves sensações de ardor e picada podem por vezes produzir-se após a instilação.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Fax: +351 21 798 73 97
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar NAABAK
Manter este medicamento fora da vista e do alcance das crianças.
Conservar a temperatura inferior a 25ºC.
Uma vez aberto, o frasco não deve ser conservado para além de 3 meses.
Não utilize este medicamento após o prazo de validade impresso na embalagem e no frasco, após "VAL". O prazo de validade corresponde ao último dia do mês indicado.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de NAABAK
- A substância ativa é o ácido N-acetil-aspartil-glutâmico (sal sódico) na concentração de 49 mg/ml.
- Os outros componentes são: hidróxido de sódio ou ácido clorídrico para ajuste do pH e água para preparações injetáveis.
Qual o aspeto de NAABAK e conteúdo da embalagem
NAABAK apresenta-se na forma de uma solução incolor e límpida, em frascos de LDPE com conta-gotas e tampa de HDPE, contendo 5 ou 10 ml de colírio.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires Théa
12 rue Louis Blériot
Zone Industrielle du Brézet
63 017 Clermont-Ferrand
França
Fabricantes
Laboratoires Thissen, S.A.
rue de la Papyrée 2-6
1420 Brain l’Alleud
Bélgica
ou
EXCELVISION
rue de la Lombardière
07100 Annonay
França
ou
FARMILA-THEA Farmaceutici S.p.A.
Via e. Fermi, 50
20019 Settimo Milanese (MI)
Itália
Este folheto foi revisto pela última vez em 04/2018

Alergia
Zaditen

Folheto informativo: Informação para o utilizador
ZADITEN 0,25 mg/ml, colírio, solução em recipiente unidose
Cetotifeno
Leia com atenção todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
- Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico, farmacêutico ou enfermeiro.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso precise de esclarecimentos ou conselhos, consulte o seu farmacêutico.
- Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Ver secção 4.
- Se não se sentir melhor ou se piorar, tem de consultar um médico.
O que contém este folheto:
- O que é Zaditen e para que é utilizado
- O que precisa de saber antes de utilizar Zaditen
- Como utilizar Zaditen
- Efeitos indesejáveis possíveis
- Como conservar Zaditen
- Conteúdo da embalagem e outras informações
1. O que é Zaditen e para que é utilizado
Zaditen contém a substância ativa cetotifeno, que é uma substância antialérgica.
Zaditen é usado para o tratamento dos sintomas oculares da “febre dos fenos”.
Se não se sentir melhor ou se piorar, tem de consultar um médico.
2. O que precisa de saber antes de utilizar Zaditen
Não utilize Zaditen:
Se tem alergia ao cetotifeno ou a qualquer outro componente deste medicamento (indicados na secção 6).
Outros medicamentos e Zaditen
Se necessita de utilizar outros colírios, deve esperar pelo menos 5 minutos entre as aplicações de cada produto.
Informe o seu médico ou farmacêutico se estiver a utilizar, tiver utilizado recentemente ou se vier a utilizar outros medicamentos.
Isto é particularmente importante no caso de medicamentos utilizados para tratar:
- depressão, ansiedade e perturbações do sono
- alergias (p. ex.: anti-histamínicos)
Zaditen com alimentos, bebidas e álcool
Zaditen pode aumentar o efeito do álcool.
Gravidez e amamentação
Se está grávida, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de utilizar este medicamento.
Zaditen pode ser utilizado durante a amamentação.
Condução de veículos e utilização de máquinas
Zaditen pode causar visão turva, sonolência ou tonturas. Se sentir algum destes sintomas, deve esperar até que desapareçam, antes de conduzir ou utilizar máquinas.
3. Como utilizar Zaditen
Utilize este medicamento exatamente como indicado pelo seu médico ou farmacêutico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico, farmacêutico ou enfermeiro. Caso precise de esclarecimentos consulte o seu médico, farmacêutico ou enfermeiro.
A dose recomendada para adultos, idosos e crianças (com 3 anos de idade ou mais), é de uma gota no(s) olho(s) afetado(s), duas vezes por dia (de manhã e à noite).
Um recipiente unidose contém solução suficiente para tratar os dois olhos na mesma utilização.
Instruções de utilização
Lave as mãos.
Abra o blister/saqueta e retire o conjunto de recipientes unidose.
Separe um dos recipientes unidose do conjunto (Fig. 1)
Coloque os restantes recipientes unidose dentro do blister/saqueta e feche dobrando o topo.
Coloque o blister/saqueta dentro da caixa.
Abra o recipiente unidose rodando a ponta. Não toque na ponta após abertura do recipiente (Fig. 2)
Incline a cabeça para trás (Fig. 3).
Puxe a pálpebra inferior ligeiramente para baixo com um dedo e segure o recipiente com a outra mão. Aperte o recipiente de modo a que caia uma gota no olho (Fig. 4).
Feche os olhos e pressione, com a ponta do dedo, o canto do olho durante 1 a 2 minutos. Desta forma irá evitar que a gota caia através do canal lacrimal para a garganta, permanecendo no olho a maior parte da gota (Fig. 5). Se necessário, repita os passos 6 a 8 para o outro olho.
Deite fora o recipiente após a utilização.

Fig. 1 Fig. 2 Fig. 3 Fig. 4 Fig. 5
Caso ainda tenha dúvidas acerca da utilização deste medicamento, fale com o seu médico, farmacêutico ou enfermeiro.
Se utilizar mais Zaditen do que deveria
Não há perigo em ingerir Zaditen acidentalmente. Da mesma forma, não se preocupe se cair mais do que uma gota no seu olho. Se tiver dúvidas, aconselhe-se junto do seu médico.
Caso se tenha esquecido de aplicar Zaditen
Se se tiver esquecido de administrar uma dose de Zaditen, aplique essa dose assim que se lembrar e retome o seu horário de dosagem regular. Não administre o dobro da dose para compensar uma dose que se esqueceu de aplicar.
4. Efeitos indesejáveis possíveis
Como todos os medicamentos, este medicamento pode causar efeitos indesejáveis, embora estes não se manifestem em todas as pessoas.
Foram notificados os seguintes efeitos indesejáveis.
Frequentes: podem afetar até 1 em 10 pessoas
- irritação ou dor ocular
- inflamação ocular
Pouco frequentes: podem afetar até 1 em 100 pessoas
- visão turva quando aplica as gotas no olho
- olho seco
- disfunção da pálpebra
- conjuntivite
- aumento da sensibilidade dos olhos à luz
- hemorragia visível na parte branca do olho
- dor de cabeça
- sonolência
- erupção cutânea (com ou sem comichão)
- eczema (erupção na pele com comichão, vermelhidão e sensação de queimadura)
- boca seca
- reação alérgica (incluindo inchaço da face e das pálpebras) e aumento da gravidade de problemas alérgicos existentes, tais como asma e eczema.
Desconhecida: a frequência não pode ser calculada com base nos dados disponíveis
-tonturas
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico ou farmacêutico. Também poderá comunicar efeitos indesejáveis diretamente ao INFARMED, I.P.:
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram (preferencialmente) ou através dos seguintes contactos:
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
E-mail: farmacovigilancia@infarmed.pt
Ao comunicar efeitos indesejáveis, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
5. Como conservar Zaditen
- Manter este medicamento fora da vista e do alcance das crianças.
- Não conservar acima de 25 ºC.
- Após abertura do blister/saqueta, os recipientes unidose podem ser armazenados durante 3 meses se estiverem dentro da caixa, ou durante 4 semanas se estiverem fora da caixa.
- O recipiente unidose não é estéril, mas o seu conteúdo é estéril até à sua abertura.
- Após a abertura do recipiente unidose, o seu conteúdo deve ser utilizado imediatamente e não deve ser guardado.
- Não utilize este após o prazo de validade impresso na embalagem e no recipiente unidose após VAL. O prazo de validade corresponde ao último dia do mês indicado.
- Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos de que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Zaditen
A substância ativa é o cetotifeno (na forma de fumarato). Cada mililitro contém 0,345 mg de fumarato de cetotifeno, correspondente a 0,25 mg de cetotifeno.
Os outros componentes são glicerol (E422), hidróxido de sódio (E524) e água para preparações injetáveis.
Qual o aspeto de Zaditen e conteúdo da embalagem
Zaditen é uma solução límpida, incolor a amarelo pálido. Cada recipiente unidose contém 0,4 ml.
Zaditen colírio em unidoses está disponível em embalagens contendo 5, 10, 20, 30, 50 e 60 recipientes unidose.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado
Laboratoires Théa
12 rue Louis Blériot
63017 Clermont-Ferrand, Cedex 2
França
Fabricante
EXCELVISION
Rue de la Lombardière
07100 Annonay
França
Ou
Laboratoire UNITHER
1 rue de l’Arquerie
50200 COUTANCES
França
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Alemanha Zaditen ophtha sine 0,25 mg/ml Augentropfen Lösung in Einzeldosisbehältnissen
Áustria Zaditen konservierungsmittelfrei 0,025% - Augentropfen in Einzeldosisbehältnissen
Dinamarca Zaditen
Espanha Zaditen colirio monodosis
Finlândia Zaditen 0,25 mg/ml silmätipat, liuos kerta-annospakkauksessa
França Zagrapa, 0,25 mg/ml, collyre en solution en récipient unidose
Grécia Zaditor οfθaλμικές staγόνeς
Holanda Zaditen Unidose 0,25 mg/ml, oogdruppels, oplossing
Islândia Zaditen
Luxemburgo Zaditen Unidose 0,25 mg/ml, collyre en solution en récipient unidose
Noruega Zaditen
Portugal Zaditen 0,25 mg/ml colírio, solução em unidoses
República Checa Zaditen SDU
Suécia Zaditen 0,25 mg/ml, ögondroppar, lösning i endosbehållare
Este folheto foi revisto pela última vez em 09/2025
Folheto informativo: Informação para o utilizador
Zaditen 0,25 mg/ml colírio, solução
Cetotifeno
Leia com atenção todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
- Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico, farmacêutico ou enfermeiro.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso precise de esclarecimentos ou conselhos, consulte o seu farmacêutico.
- Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Ver secção 4.
- Se não se sentir melhor ou se piorar, tem de consultar um médico.
O que contém este folheto:
1. O que é Zaditen e para que é utilizado
2. O que precisa de saber antes de utilizar Zaditen
3. Como utilizar Zaditen
4. Efeitos indesejáveis possíveis
5. Como conservar Zaditen
6. Conteúdo da embalagem e outras informações
1. O que é Zaditen e para que é utilizado
Zaditen contém a substância ativa cetotifeno, que é uma substância antialérgica.
Zaditen é usado para o tratamento dos sintomas oculares da “febre dos fenos”.
Se não se sentir melhor ou se piorar após 7 dias, tem de consultar um médico.
2. O que precisa de saber antes de utilizar Zaditen
Não utilize Zaditen
Se tem alergia ao cetotifeno ou a qualquer outro componente deste medicamento (indicados na secção 6).
Advertências e precauções
Fale com o seu médico, farmacêutico ou enfermeiro antes de utilizar Zaditen
Outros medicamentos e Zaditen
Se necessita de utilizar outros colírios conjuntamente com Zaditen, deve esperar pelo menos 5 minutos entre as aplicações de cada produto.
Informe o seu médico ou farmacêutico se estiver a utilizar, tiver utilizado recentemente ou se vier a utilizar outros medicamentos.
Isto é particularmente importante no caso de medicamentos utilizados para tratar:
- depressão, ansiedade e perturbações do sono
- alergias (por exemplo anti-histamínicos)
Zaditen com alimentos, bebidas e álcool
Zaditen pode aumentar o efeito do álcool
Gravidez e amamentação
Se está grávida, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de utilizar este medicamento.
Zaditen pode ser utilizado durante a amamentação.
Condução de veículos e utilização de máquinas
Zaditen pode causar visão turva, sonolência ou tonturas. Se sentir algum destes sintomas deve esperar que desapareçam antes de conduzir ou utilizar máquinas.
Zaditen contém cloreto de benzalcónio
Este medicamento contém 2,6 microgramas de cloreto de benzalcónio em cada gota.
O cloreto de benzalcónio pode ser absorvido pelas lentes de contacto moles e pode alterar a cor das lentes de contacto. Deve remover as lentes de contacto antes da utilização deste medicamento e voltar a colocá-las 15 minutos depois.
O cloreto de benzalcónio pode também causar irritação nos olhos, especialmente se tem os olhos secos ou alterações da córnea (a camada transparente na parte da frente do olho). Se tiver uma sensação estranha no olho, picadas ou dor no olho após utilizar este medicamento, fale com o seu médico.
3. Como utilizar Zaditen
Utilize este medicamento exatamente como indicado pelo seu médico ou farmacêutico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico, farmacêutico ou enfermeiro. Caso precise de esclarecimentos consulte o seu médico, farmacêutico ou enfermeiro.
A dose recomendada para adultos, idosos e crianças (com 3 anos de idade ou mais) é de uma gota no(s) olho(s) afetado(s) duas vezes por dia (de manhã e à noite).
Instruções de utilização
1. Lave as mãos.
2. Abra o frasco. Não toque na ponta do frasco depois de aberto.
3. Incline a cabeça para trás (Fig. 1).
4. Puxe a pálpebra inferior ligeiramente para baixo com um dedo e segure o frasco com a outra mão. Aperte o frasco de modo a que caia uma gota no olho (Fig. 2).
5. Feche os olhos e pressione, com a ponta do dedo, o canto do olho durante 1 a 2 minutos. Desta forma irá evitar que a gota caia através do canal lacrimal para a garganta, permanecendo no olho a maior parte da gota (Fig. 3). Se necessário, repita os passos 3 a 5 para o outro olho.
6. Feche o frasco após a utilização.

Fig. 1 Fig. 2 Fig. 3
Caso ainda tenha dúvidas acerca da utilização deste medicamento, fale com o seu médico, farmacêutico ou enfermeiro.
Se utilizar mais Zaditen do que deveria
Não há perigo em ingerir Zaditen acidentalmente. Da mesma forma, não se preocupe se cair mais do que uma gota no seu olho. Se tiver dúvidas, aconselhe-se junto do seu médico.
Caso se tenha esquecido de aplicar Zaditen
Se se tiver esquecido de administrar uma dose de Zaditen, aplique essa dose assim que se lembrar e retome o seu horário de dosagem regular. Não administre o dobro da dose para compensar uma dose que se esqueceu de aplicar.
4. Efeitos indesejáveis possíveis
Como todos os medicamentos, este medicamento pode causar efeitos indesejáveis, embora estes não se manifestem em todas as pessoas.
Foram notificados os seguintes efeitos indesejáveis:
Frequentes: podem afetar até 1 em 10 pessoas
- irritação ou dor ocular
- inflamação ocular
Pouco frequentes: podem afetar até 1 em 100 pessoas
- visão turva quando as gotas são administradas no olho
- olho seco
- disfunção da pálpebra
- conjuntivite
- aumento da sensibilidade dos olhos à luz
- hemorragia visível na parte branca do olho
- dor de cabeça
- sonolência
- erupção cutânea (com ou sem comichão)
- eczema (erupção na pele com comichão, vermelhidão e sensação de queimadura)
- boca seca
- reação alérgica (incluindo inchaço da face e das pálpebras) e aumento da gravidade de condições alérgicas pré-existentes tais como asma e eczema.
Desconhecida: a frequência não pode ser calculada com base nos dados disponíveis
- tonturas
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico ou farmacêutico. Também poderá comunicar efeitos indesejáveis diretamente ao INFARMED, I.P. através dos contactos abaixo:
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram (preferencialmente) ou através dos seguintes contactos:
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Fax: +351 21 798 73 97
E-mail: farmacovigilancia@infarmed.pt
Ao comunicar efeitos indesejáveis, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
5. Como conservar Zaditen
Manter este medicamento fora da vista e do alcance das crianças.
Não conservar acima de 25ºC.
O frasco não é estéril, mas o seu conteúdo é estéril até à abertura do frasco.
Após a abertura do frasco, o colírio pode ser armazenado durante apenas 4 semanas.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior e no frasco após VAL. O prazo de validade corresponde ao último dia do mês indicado.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Zaditen
A substância ativa é o cetotifeno (na forma de fumarato). Cada mililitro contém 0,345 mg de fumarato de cetotifeno, correspondente a 0,25 mg de cetotifeno.
Os outros componentes são glicerol (E422), hidróxido de sódio (E524), água para preparações injetáveis e cloreto de benzalcónio.
Qual o aspeto de Zaditen e conteúdo da embalagem
Zaditen é uma solução límpida, incolor a amarelo pálido. A solução está disponível numa embalagem contendo um frasco de 5 ml.
Titular da Autorização de Introdução no Mercado
Laboratoires THEA
12, rue Louis Blériot
63017 Clermont-Ferrand Cedex - França
Fabricante
EXCELVISION
Rue de la Lombardière
07100 Annonay
França
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Áustria Zaditen 0,025% - Augentropfen
Dinamarca Zaditen
Finlândia Zaditen 0,25 mg/ml silmätipat, liuos
Alemanha Zaditen ophtha 0,25 mg/ml Augentropfen
Grécia Zaditen οfθaλμικές staγόνeς
Islândia Zaditen
Irlanda Zaditen 0.25mg/ml, eye drops, solution
Luxemburgo Zaditen 0,25 mg/ml, collyre en solution
Noruega Zaditen
Portugal Zaditen 0,25 mg/ml colírio, solução
Espanha Zaditen colirio
Suécia Zaditen 0,25 mg/ml, ögondroppar, lösning
Holanda Zaditen 0,25 mg/ml, oogdruppels, oplossing
Reino Unido Zaditen 0.25 mg/ml, eye drops, solution
Este folheto foi revisto pela última vez em 09/2025

Alergia
Zabak
Folheto informativo: Informação para o utilizador
ZABAK 0,25 mg/ml, colírio, solução
Cetotifeno
Leia com atenção todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico ou farmacêutico.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso precise de esclarecimentos ou conselhos, consulte o seu farmacêutico.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico farmacêutico. Ver secção 4.
- Se não se sentir melhor ou se piorar, tem de consultar um médico.
O que contém este folheto:
1. O que é ZABAK e para que é utilizado
2. O que precisa de saber antes de utilizar ZABAK
3. Como utilizar ZABAK
4. Efeitos secundários possíveis
5. Como conservar ZABAK
6. Conteúdo da embalagem e outras informações
1. O que é ZABAK e para que é utilizado
Zabak é um colírio, solução sem conservantes, contendo cetotifeno, que é um agente antialérgico.
Zabak é utilizado no tratamento dos sintomas da conjuntivite alérgica sazonal.
Os sintomas mais comuns da conjuntivite alérgica sazonal são a comichão intensa em ambos os olhos, associada a vermelhidão, inchaço, lacrimejo excessivo, inflamação e sensibilidade à luz. Os sintomas são recorrentes e desencadeados pelo início da temporada do pólen. A conjuntivite alérgica sazonal também pode estar associada a outros sintomas da febre dos fenos, como a rinite.
Se não se sentir melhor ou se piorar após 7 dias, tem de consultar um médico (ver secção 2).
2. O que precisa de saber antes de utilizar ZABAK
Não utilize Zabak
- Se tem alergia (hipersensibilidade) ao cetotifeno (a substância ativa), ou a qualquer outro componente deste medicamento (indicados na secção 6).
Advertências e precauções
Fale com o seu médico, farmacêutico ou enfermeiro antes de utilizar ZABAK.
Deve falar com um médico se não se sentir melhor após 7 dias ou se se sentir pior.
Os seguintes sinais e sintomas não são habituais na conjuntivite alérgica sazonal:
- secreção purulenta,
- diminuição da visão,
- crosta matinal, dificuldade em abrir as pálpebras de manhã,
- nódulos linfáticos inchados perto das orelhas,
- sintomas persistentes,
- sintomas unilaterais.
Se sentir um destes sinais ou sintomas, pode sofrer de outra condição ocular para além conjuntivite alérgica sazonal. Deve procurar aconselhamento médico.
Outros medicamentos e ZABAK
Se necessita de aplicar qualquer outro medicamento nos seus olhos em conjunto com Zabak, espere pelo menos 5 minutos entre as doses.
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente outros medicamentos.
Tal é particularmente importante no caso de medicamentos utilizados no tratamento da:
- depressão, ansiedade e perturbações do sono,
- alergia (por exemplo, anti-histamínico).
ZABAK com alimentos, bebidas e álcool
- Zabak pode aumentar o efeito do álcool.
Gravidez e amamentação
É preferível evitar a utilização de Zabak durante a gravidez.
Se está grávida, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento
Zabak pode ser utilizado durante a amamentação.
Condução de veículos e utilização de máquinas
Zabak pode causar visão turva ou sonolência.
Se tal lhe acontecer, espere até que estes efeitos desapareçam antes de conduzir ou utilizar máquinas.
3. Como utilizar ZABAK
Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico, farmacêutico ou enfermeiro. Fale com o seu médico, farmacêutico ou enfermeiro se tiver dúvidas.
Posologia
Adultos, idosos e crianças com idade superior a 3 anos
A dose habitual é de uma gota no olho ou nos olhos afetados, duas vezes por dia (de manhã e à noite).
Modo de administração
Não injetar, não engolir.
Este medicamento destina-se a ser aplicado no olho (uso oftálmico).
Antes de utilizar pela primeira vez, rejeite as 5 primeiras gotas.
Depois, cada vez que utilizar o medicamento:
1- Lave cuidadosamente as mãos antes de utilizar o medicamento.
2- Para evitar a contaminação das gotas, não toque com a extremidade do conta-gotas no olho ou nas pálpebras ou em qualquer superfície.
3- Aplique uma gota no(s) olho(s), olhando para cima e puxando suavemente a pálpebra inferior para baixo.
Feche a pálpebra e pressione suavemente o canto interno do olho com o dedo indicador por 1-2 minutos. Tal ajudará a evitar que o medicamento se espalhe para o resto do corpo.
4- Feche o frasco após a utilização.
Se pensa que os efeitos do Zabak são demasiado fortes ou demasiado fracos, ou se sentir quaisquer reações indesejadas, contacte o seu médico ou farmacêutico.
Se utilizar mais ZABAK do que deveria
Não há perigo se ingerir acidentalmente algumas gotas de Zabak. De igual modo, não se preocupe se aplicar acidentalmente mais do que uma gota no seu olho.
Caso se tenha esquecido de utilizar ZABAK
Aplique uma gota logo que possível e depois retome o seu esquema habitual de administração. Certifique-se que a gota é aplicada no seu olho. Não administre uma dose a dobrar para compensar uma dose de que se esqueceu.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos indesejáveis possíveis
Como todos os medicamentos, este medicamento pode causar efeitos indesejáveis, embora estes não se manifestem em todas as pessoas.
Se sofrer de reações alérgicas, incluindo erupção cutânea, inchaço do rosto, lábios, língua e/ou garganta, que podem causar dificuldade em respirar ou engolir, ou se ocorrerem outros efeitos indesejáveis graves, deve parar de utilizar Zabak e entrar em contato imediatamente com seu médico ou com as urgências do hospital mais próximo.
Foram notificados os seguintes efeitos indesejáveis:
Frequentes (afetando menos de 1 em cada 10 doentes):
- irritação ou dor ocular,
- inflamação no olho.
Pouco frequentes (afetando menos de 1 em cada 100 doentes):
- visão turva,
- olho seco,
- afeção da pálpebra,
- conjuntivite (inflamação da superfície do olho),
- aumento da sensibilidade dos olhos à luz,
- hemorragia visível na parte branca do olho,
- dor de cabeça,
- sonolência,
- erupção cutânea (que também pode dar comichão),
- eczema (erupção com comichão, vermelhidão e sensação de queimadura),
- boca seca.
Desconhecido: a frequência não pode ser calculada a partir dos dados disponíveis:
- reação alérgica (incluindo inchaço da face e pálpebras) e aumento da gravidade das condições alérgicas existentes, como asma e eczema,
- tonturas
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos indesejáveis diretamente através dos contactos abaixo. Ao comunicar efeitos indesejáveis, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
(preferencialmente)
ou através dos seguintes contactos:Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
E-mail: farmacovigilancia@infarmed.pt
As reações adversas também podem ser comunicadas ao Titular de Autorização de Introdução no Mercado.
5. Como conservar ZABAK
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na cartonagem e no frasco após EXP. O prazo de validade corresponde ao último dia do mês indicado.
Zabak pode ser utilizado até 3 meses após a primeira abertura do frasco.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos de que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Zabak
- A substância ativa é:
Hidrogenofumarato de cetotifeno……………….…0,345 mg
sob a forma de cetotifeno …………………………..0,25 mg
para 1 ml
- Os outros componentes são glicerol, hidróxido de sódio (para ajuste do pH), água para preparações injetáveis.
Qual o aspeto de Zabak e conteúdo da embalagem
Zabak apresenta-se sob a forma de colírio, solução em frasco de 5 ml multidose (no mínimo 150 gotas sem conservante). A solução é incolor ou ligeiramente acastanhada/amarelada.
Titular da Autorização de Introdução no Mercado
LABORATOIRES THEA
12, rue Louis Blériot
63017 CLERMONT-FERRAND CEDEX 2
FRANÇA
Fabricante
EXCELVISION,
rue de la Lombardière
07100 ANNONAY
FRANÇA
ou
FARMILA-THEA Farmaceutici S.p.A
Via E. Fermi, 50
20019 Settimo Milanese (MI)
ITALY
ou
LABORATOIRES THEA
12, rue Louis Blériot
63017 CLERMONT-FERRAND CEDEX 2
FRANÇA
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu sob as seguintes denominações:
Áustria .................................................................................. ZADITEN OPHTHA ABAK
Itália.................................................................................................ZADITEN OFTABAK
Bulgária, Grécia, Polónia, Portugal, Roménia……………………ZABAK
Dinamarca, França, Espanha ……………………………………………ZALERG
Bélgica, Luxemburgo, Holanda…………………………………………ALTRIABAK
Este folheto foi revisto pela última vez em 10/2021
- Anti-virais

Anti-virais
Virgan

Folheto informativo: Informação para o utilizador
Virgan Gele Oftálmico 1,5 mg/g
Ganciclovir
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois
contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico. Ver secção 4.
O que contém este folheto:
- O que é Virgan Gele Oftálmicoe para que é utilizado
- O que precisa de saber antes de utilizar Virgan Gele Oftálmico
- Como utilizar Virgan Gele Oftálmico
- Efeitos secundários possíveis
- Como conservar Virgan Gele Oftálmico
- Conteúdo da embalagem e outras informações
1. O que é Virgan Gele Oftálmico e para que é utilizado
Virgan Gele Oftálmico é um gel oftálmico que contém um agente antiviral denominado ganciclovir.
Está indicado no tratamento de determinadas infeções virais superficiais do olho (córnea).
2. O que precisa de saber antes de utilizar Virgan Gele Oftálmico
Não utilize Virgan Gele Oftálmico
- se tem alergia ao ganciclovir ou a qualquer outro componente deste medicamento (indicados na secção 6).
- durante a gravidez e o aleitamento, exceto se indicado pelo seu médico.
- em homens e mulheres com potencial para engravidar, que não utilizem um método contracetivo eficaz.
Adicionalmente, os homens tratados com Virgan devem ser alertados para utilizarem um método contracetivo local (como, por exemplo, preservativo) durante o tratamento e até três meses após o final do tratamento. As mulheres tratadas com Virgan devem ser aconselhadas a utilizar contracetivos durante o tratamento e até seis meses após o final do tratamento.
Advertências e precauções
Fale com o seu médico ou farmacêutico antes de utilizar Virgan Gele Oftálmico
- não é recomendada a utilização em crianças com menos de 18 anos, pois não existe nenhum estudo específico.
- não engolir.
- manter fora do alcance e da vista das crianças.
- evitar tocar no olho ou nas pálpebras com a ponta do conta-gotas.
- o cloreto de benzalcónio pode causar irritação ocular.
- uso de lentes de contacto: evitar o contacto com lentes de contacto hidrófilas. Retirar as lentes de contacto antes da aplicação e esperar pelo menos 15 minutos antes de as voltar a colocar.
Virgan Gele Oftálmico contém cloreto de benzalcónio que pode descolorar as lentes de contacto.
Outros medicamentos e Virgan Gele Oftálmico
Informe o seu médico ou farmacêutico se estiver a utilizar, ou tiver utilizado recentemente, ou se vier a utilizar outros medicamentos.
Se utiliza qualquer outro medicamento de aplicação ocular, deve:
- aplicar o outro medicamento oftálmico,
- esperar durante 15 minutos,
- aplicar Virgan Gele Oftálmico em último lugar.
Gravidez e amamentação
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento.
Este medicamento não deve ser utilizado durante a gravidez e o aleitamento, exceto se indicado pelo seu médico.
Consulte imediatamente o seu médico se pretende ficar grávida ou se descobrir que está grávida durante o tratamento. O seu médico é o único que pode ajustar o seu tratamento.
Consulte o seu médico ou farmacêutico antes de tomar qualquer medicamento.
Condução de veículos e utilização de máquinas
Pode sentir perturbações visuais temporárias após a administração ocular deste medicamento.
Aguarde até que tenha recuperado a sua visão normal antes de conduzir veículos ou utilizar máquinas.
Virgan Gele Oftálmico contém cloreto de benzalcónio
Ver secção 2: Advertências e precauções.
3. Como utilizar Virgan Gele Oftálmico
Utilize este medicamento exatamente como indicado pelo seu médico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
A dose recomendada é de 1 gota, 3 a 5 vezes ao dia.
Utilização em crianças
Não é recomendada a utilização deste gel oftálmico em crianças com menos de 18 anos.
Modo de administração
Este medicamento destina-se a ser aplicado no olho (uso ocular).
- Lave cuidadosamente as suas mãos antes de aplicar o produto,
- Pressione suavemente a bisnaga para libertar uma gota no olho afetado, enquanto olha para cima e puxa a pálpebra inferior para baixo.
- Feche a bisnaga após a utilização.
Duração do tratamento
Normalmente o tratamento não deve exceder 21 dias.
Se utilizar mais Virgan Gele Oftálmico do que deveria
Continue o seu tratamento como indicado pelo seu médico.
Contacte imediatamente o seu médico ou farmacêutico.
Caso se tenha esquecido de utilizar Virgan Gele Oftálmico
Não utilize uma dose a dobrar para compensar uma dose que se esqueceu de utilizar.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes
não se manifestem em todas as pessoas.
Afeções oculares:
Muito frequentes (em mais de 1 em 10 doentes):
- Sensações transitórias de ardor ou picada.
Frequentes (em menos de 1 em 10 doentes, mas em mais de 1 em 100 doentes):
- Inflamação da córnea (queratite ponteada superficial).
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico. Também poderá comunicar efeitos secundários
diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 71 40
Fax: +351 21 798 73 97
Sítio da internet: http://extranet.infarmed.pt/page.seram.frontoffice.seramhomepage
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar VIRGAN Gele Oftálmico
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na bisnaga e na embalagem exterior após VAL. O prazo de validade corresponde ao último dia do mês indicado.
Não conservar acima de 25ºC.
A bisnaga não deve ser utilizada mais de 4 semanas após a sua abertura.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu
farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a
proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Virgan Gele Oftálmico
A substância ativa é o ganciclovir. Cada grama de gel contém 1,5 mg de ganciclovir.
Os outros componentes são carbómero (carbopol 974P), sorbitol, hidróxido de sódio, cloreto de benzalcónio e água para preparações injetáveis.
Qual o aspeto de Virgan Gele Oftálmico e conteúdo da embalagem
Este medicamento apresenta-se numa bisnaga contendo 5 g de gel oftálmico.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
LABORATOIRES THEA
12, rue Louis Blériot - Zone Industrielle du Brézet
63017 CLERMONT-FERRAND Cedex 2
FRANÇA
Fabricante
FARMILA-THEA FARMACEUTICI Spa
Via Enrico Fermi, 50
SETTIMO MILANESE (MI)
ITÁLIA
Este folheto foi revisto pela última vez em 06/2023
- Antibióticos

Antibióticos
Azyter

Folheto informativo: Informação para o utilizador
Azyter 15 mg/g colírio, solução em recipiente unidose
Azitromicina di-hidratada
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico ou com o médico do seu filho, ou farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si ou para o seu filho. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico ou enfermeiro.
O que contém este folheto:
- O que é AZYTER e para que é utilizado
- O que precisa de saber antes de utilizar AZYTER
- Como utilizar AZYTER
- Efeitos secundários possíveis
- Como conservar AZYTER
- Conteúdo da embalagem e outras informações
1. O que é AZYTER e para que é utilizado
Azyter contém azitromicina, um antibiótico pertencente à classe dos macrólidos.
Azyter é utilizado no tratamento localizado de certas infeções oculares de origem bacteriana em adultos (incluindo idosos) e crianças desde o nascimento até aos 17 anos de idade.
- conjuntivites bacterianas purulentas,
- conjuntivites do tracoma (infeção ocular particular devida a uma bactéria denominada Chlamydia trachomatis, que existe ou aparece em certos países em vias de desenvolvimento).
2. O que precisa de saber antes de utilizar AZYTER
Não utilize Azyter
Se tem alergia à azitromicina, a qualquer outro antibiótico da classe dos macrólidos ou aos triglicéridos de cadeia média.
Advertências e precauções
Fale com o seu médico ou com o médico do seu filho ou com o seu farmacêutico ou enfermeiro antes de utilizar AZYTER.
- Se ocorrer uma reação alérgica, deverá interromper o tratamento e consultar o seu médico.
- Caso não observe nenhuma melhoria no espaço de três dias após o início do tratamento ou se ocorrerem sinais anómalos, deverá consultar o seu médico.
- Devido à sua infeção ocular, não se recomenda a utilização de lentes de contacto.
Este medicamento destina-se apenas a uso oftálmico.
Não injete ou ingira.
Outros medicamentos e AZYTER
Se utiliza qualquer outro medicamento para aplicar no olho, deverá:
1- aplicar o outro medicamento oftálmico,
2- esperar durante 15 minutos,
3- aplicar o Azyter por último.
Informe o seu médico ou o médico do seu filho ou o farmacêutico se estiver a utilizar, tiver utilizado recentemente, ou se vier a utilizar outros medicamentos.
Gravidez
Azyter pode ser usado durante a gravidez.
O seu médico poderá prescrever-lhe este medicamento se estiver grávida, caso considere necessário.
Amamentação
Dados limitados indicam que a azitromicina é excretada no leite materno. Este medicamento pode ser usado durante amamentação.
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de utilizar este medicamento.
Condução de veículos e utilização de máquinas
Poderá sentir uma turvação temporária da visão após a administração do produto no olho.
Espere até recuperar a visão normal antes de conduzir um veículo ou utilizar qualquer máquina.
3. Como utilizar AZYTER
Este medicamento destina-se a ser administrado no olho (uso oftálmico).
Utilize este medicamento exatamente como indicado pelo seu médico ou pelo médico do seu filho. Fale com o seu médico, com o médico do seu filho ou farmacêutico se tiver dúvidas.
Posologia
A dose recomendada para adultos (incluindo idosos) e crianças desde o nascimento até aos 17 anos de idade é de uma gota duas vezes por dia no(s) olhos(s) a ser(em) tratado(s): uma gota de manhã e uma gota à noite.
A duração do tratamento é de três dias. Dada a ação prolongada do produto, não é necessário continuar o tratamento para além dos três dias, mesmo que ainda apresente sinais residuais da sua infeção bacteriana.
Modo de administração
Para uma correta administração do Azyter:
- Lave cuidadosamente as mãos antes e depois de utilizar o produto,
- Coloque uma gota no olho a ser tratado, olhando para cima e puxando ligeiramente a pálpebra inferior para baixo,
- Evite tocar no olho e nas pálpebras com o conta-gotas do recipiente unidose,
- Rejeite o recipiente unidose após a utilização. Não o guarde para uma utilização subsequente.
NÃO INJETE, NÃO INGIRA
Caso se tenha esquecido de utilizar Azyter
Não utilize uma dose a dobrar para compensar uma dose que se esqueceu de utilizar.
Se parar de utilizar Azyter
Consulte sempre o seu médico antes de considerar interromper o tratamento.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico ou enfermeiro.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Informe imediatamente o seu médico se ocorrer algum dos seguintes sintomas após a utilização deste medicamento uma vez que estes podem ser graves.
Pouco frequentes: podem afetar 1 em 100 pessoas:
- reação alérgica grave que provoca inchaço da face ou da garganta (angioedema).
Desconhecida: a frequência não pode ser calculada a partir dos dados disponíveis
- reações na pele graves: exantema na pele, erupções na pele, bolhas na pele, boca, olhos e genitais (Síndrome de Stevens-Johnson (SSJ) ou Necrólise Epidérmica Tóxica (NET)), exantema acompanhado por outros sintomas tais como febre, inchaço das glândulas e aumento dos eosinófilos (um tipo de glóbulos brancos), uma forma de exantema com inchaços pequenos, vermelhos com comichão (Reação ao fármaco com eosinofilia e sintomas sistémicos (DRESS)), eritema generalizado com descamação em extensas áreas do corpo (Dermatite Exfoliativa), erupção na pele caracterizada pelo rápido aparecimento de zonas vermelhas na pele com pequenas pústulas (pequenas bolhas com líquido branco/amarelo) (Pustulose Generalizada Exantemática Aguda (PGEA)).
Pare de utilizar azitromicina se desenvolver este tipo de sintomas na pele e contacte o seu médico ou procure assistência médica imediatamente.
Após a administração do produto, foram observados os seguintes efeitos secundários:
Muito frequentes: podem afetar mais de 1 em 10 pessoas:
- desconforto ocular transitório (comichão, ardor, picadas).
Frequentes: podem afetar até 1 em 10 pessoas:
- turvação da visão,
- sensação de olho colado,
- sensação de corpo estranho.
Pouco frequentes: podem afetar 1 em 100 pessoas:
- reação alérgica (hipersensibilidade),
- inflamação da conjuntiva (pode ser devida a infeção ou alergia) (conjuntivite),
- inflamação alérgica da conjuntiva (conjuntivite alérgica),
- inflamação da córnea (queratite),
- comichão na pele das pálpebras (eczema das pálpebras),
- secura, vermelhidão, inchaço das pálpebras (edema da pálpebra),
- alergia no olho,
- lacrimejo,
- vermelhidão das pálpebras (eritema das pálpebras),
- vermelhidão da conjuntiva (hiperemia conjuntival).
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, o médico do seu filho, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contatos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar AZYTER
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior e no recipiente unidose. O prazo de validade corresponde ao último dia do mês indicado.
- Não conservar acima de 25ºC.
- Manter os recipientes unidose dentro da saqueta para os proteger da luz.
Os recipientes unidose abertos e com qualquer solução que ainda reste, devem ser imediatamente rejeitados após a primeira utilização. Não os guarde para uma utilização subsequente.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Azyter
- A substância ativa é a azitromicina di-hidratada. Cada grama de solução contém 15 mg de azitromicina di-hidratada equivalentes a 14,3 mg de azitromicina. Cada recipiente unidose de 250 mg de solução contém 3,75 miligramas de azitromicina di-hidratada.
- Os outros componentes são os triglicéridos de cadeia média.
Qual o aspeto de Azyter e conteúdo da embalagem
Azyter é uma solução para uso oftálmico (colírio, solução) apresentado em recipientes unidose, contendo cada 0,25 g de medicamento. Trata-se de um líquido oleoso, límpido, incolor a ligeiramente amarelado.
A embalagem contém seis recipientes unidose numa saqueta, suficientes para um tratamento.
Titular da Autorização de Introdução no Mercado
LABORATOIRES THEA
12, rue Louis Blériot
63017 CLERMONT-FERRAND CEDEX 2
FRANÇA
Fabricante
LABORATOIRES THEA
12, rue Louis Blériot
63017 CLERMONT-FERRAND CEDEX 2
FRANÇA
ou
Laboratoire UNITHER
ZI de la Guérie
50200 COUTANCES
FRANÇA
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Alemanha, Áustria, Bulgária, Chipre, Dinamarca, Eslovénia, Finlândia, França, Grécia, Holanda, Irlanda, Itália, Islândia, Luxemburgo, Noruega, Polónia, Portugal, Reino Unido, República Checa, Roménia, Suécia…….AZYTER
Espanha…………AZYDROP
Este folheto foi revisto pela última vez em 11/2021
- Cirurgia

Cirurgia
Mydriasert

Folheto informativo: Informações para o utilizador
MYDRIASERT 5.4 mg + 0.28 mg Inserto oftálmico
Cloridrato de fenilefrina + tropicamida
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico, farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
1. O que é MYDRIASERT e para que é utilizado
2. O que precisa de saber antes de utilizar MYDRIASERT
3. Como utilizar MYDRIASERT
4. Efeitos indesejáveis possíveis
5. Como conservar MYDRIASERT
6. Conteúdo da embalagem e outras informações
1. O que é MYDRIASERT e para que é utilizado
MYDRIASERT é um medicamento oftálmico, isto é, exclusivamente para tratamento ocular.
A utilização de MYDRIASERT é destinada apenas a profissionais de saúde.
MYDRIASERT destina-se a ser inserido pelo pessoal médico na pálpebra inferior do seu olho. Utiliza-se para obter uma midríase (dilatação da pupila), antes de uma intervenção cirúrgica ou para fins de diagnóstico.
2. O que precisa de saber antes de utilizar MYDRIASERT
NÃO UTILIZE MYDRIASERT nos seguintes casos:
- Se tem alergia à tropicamida ou ao cloridrato de fenilefrina ou a qualquer outro componente deste medicamente (indicados na secção 6). - Em doentes com glaucoma de ângulo fechado ou em risco de precipitação de glaucoma (aumento da pressão ocular).
- Em crianças com idade inferior a 12 anos.
Advertências e precauções
- Uma vez que este medicamento causa perturbações visuais prolongadas, tenha o cuidado de vir acompanhado à consulta (ver efeitos indesejáveis possíveis).
- Em caso de desconforto após inserção do inserto, informe o seu médico: pode ocorrer uma deslocação ou, mais raramente, a perda do inserto.
- Se sofre de grave secura dos olhos, o médico poderá ter de colocar uma gota de solução salina no olho para reduzir o risco de irritação ocular.
- Em caso de hipertensão (pressão arterial elevada), aterosclerose (espessamento da parede arterial), doença cardíaca ou hipertiroidismo (aumento da atividade da glândula tiroide) ou perturbações prostáticas, informe o seu médico.
- Em determinadas pessoas com predisposição, os agentes midriáticos (produtos que dilatam a pupila) podem desencadear uma crise de glaucoma agudo (devido ao súbito aumento da pressão ocular).
- É desaconselhado o uso de lentes de contacto hidrófilas durante o tratamento.
Crianças e adolescentes
Mydriasert não deve ser utilizado em crianças com idade inferior a 12 anos, uma vez que as crianças parecem ser mais sensíveis ao risco de efeitos indesejáveis graves. Não se recomenda a utilização de Mydriasert em crianças com idades entre os 12 e os 18 anos pois não existe experiência clínica adequada.
Outros medicamentos e MYDRIASERT
Informe o seu médico se estiver a usar medicamentos que dilatem as pupilas (midríase), para além de Mydriasert, de forma que o médico possa determinar a quantidade total de agente midriático à qual ficará exposto.
Informe o seu médico ou farmacêutico se estiver a tomar, tiver tomado recentemente ou se vier a tomar outros medicamentos, incluindo medicamentos obtidos sem receita médica. Embora Mydriasert seja aplicado localmente, estas informações são importantes, uma vez que alguns medicamentos podem afetar a ação de outros.
Gravidez e aleitamento
Não existem dados adequados sobre a utilização de Mydriasert ou das suas substâncias ativas em mulheres grávidas. Por isso, Mydriasert não deverá ser utilizado durante a gravidez, a menos que seja claramente necessário.
A utilização durante o aleitamento não é recomendada.
Condução de veículos e utilização de máquinas
Não conduza ou utilize máquinas porque MYDRIASERT pode causar perturbações visuais incómodas por algumas horas (turvação devido à dilatação prolongada da pupila).
Informações importantes sobre alguns componentes de MYDRIASERT
Os desportistas devem ser alertados para o facto de este medicamento conter uma substância ativa (o cloridrato de fenilefrina) que pode originar resultados positivos nos testes para deteção de substâncias proibidas.
3. Como utilizar MYDRIASERT
DESTINADO APENAS A UTILIZAÇÃO POR PROFISSIONAIS DE SAÚDE.
Mydriasert destina-se a ser usado em adultos. Mydriasert não deve ser ingerido.
O profissional de saúde colocará um inserto oftálmico dentro da pálpebra inferior do olho em questão. O profissional retirará o inserto em 30 minutos, quando a pupila se apresentar suficientemente dilatada, e antes de proceder à operação ou à investigação do olho. O inserto oftálmico não deve permanecer dentro do olho por mais de 2 horas.
Se utilizar mais MYDRIASERT do que deveria
Dado que um único inserto será inserido no olho pelo médico ou profissional de saúde, é improvável o risco de utilizar mais Mydriasert do que o recomendado. No entanto, se o profissional de saúde precisar de usar gotas oculares midriáticas, para além de Mydriasert, pode ocorrer o risco de sobredosagem das substâncias ativas de Mydriasert.
Os sintomas de sobredosagem das substâncias ativas de Mydriasert podem incluir fadiga extrema, sudação, tonturas, diminuição do ritmo cardíaco, coma, dores de cabeça, aumento do ritmo cardíaco, boca e pele seca, sonolência invulgar, rubor e dilatação prolongada das pupilas.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Frequentes: podem afetar até 1 em 10 pessoas:
- picadas,
- visão enevoada,
- desconforto visual devido à perceção da presença ou deslocação do inserto oftálmico.
Pouco frequentes: podem afetar até 1 em 100 pessoas:
- lacrimejo,
- irritação,
- encadeamento devido à dilatação prolongada da pupila,
- queratite ponteada superficial (inflamação da córnea).
Raros: podem afetar até 1 em 1.000 pessoas:
- reações alérgicas: inflamação das pálpebras (blefarite), inflamação da conjuntiva (conjuntivite).
Muito raros: podem afetar até 1 em 10.000 pessoas:
- convulsões.
Foram observadas úlceras da córnea (pequenas erosões da superfície do olho) e edema da córnea (inflamação da superfície do olho) quando o inserto oftálmico foi acidentalmente deixado dentro do olho.
Em indivíduos com predisposição, MYDRIASERT pode desencadear uma crise de glaucoma agudo (súbito aumento da pressão intraocular):em caso de sintomas anormais após a administração (vermelhidão, dor e perturbações visuais), contacte imediatamente o seu médico.
Embora improvável após administração no olho, as substâncias ativas contidas em MYDRIASERT podem causar os seguintes efeitos secundários, que deverão ser tidos em consideração:
- aumento da pressão arterial, taquicardia,
- muito raramente, incidentes mais graves, como arritmia cardíaca,
- tremores, palidez, dores de cabeça, boca seca.
Efeitos indesejáveis adicionais em crianças
Frequência desconhecida (não pode ser calculada a partir dos dados disponíveis)
- Fluido ou inchaço nos pulmões
- Palidez em redor dos olhos em bebés prematuros
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar MYDRIASERT
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na saqueta após "VAL".
Não conservar acima de 25°C.
Usar imediatamente após abertura da saqueta.
Não utilizar MYDRIASERT se observar qualquer deterioração no invólucro da saqueta ou no inserto oftálmico.
6. Conteúdo da embalagem e outras informações
Qual a composição de MYDRIASERT
- As substâncias ativas são tropicamida 0.28 mg e cloridrato de fenilefrina 5.4 mg, em cada inserto oftálmico.
- Os outros componentes são o metacrilato de amónio copolímero tipo A, Eudragit NE 30 D, dibehenato de glicerilo e etilcelulose.
Qual o aspeto de MYDRIASERT e conteúdo da embalagem
MYDRIASERT é fornecido numa saqueta.
MYDRIASERT tem o formato de uma pequena placa (inserto) branca, oblonga (4.3 mm x 2.3 mm).
Cada embalagem contém outra saqueta com uma pinça esterilizada descartável, usada para colocação de MYDRIASERT no olho.
Embalagens de 1 inserto e 1 pinça, 10 insertos e 10 pinças, 20 insertos e 20 pinças, 50 insertos e 50 pinças ou 100 insertos e 100 pinças.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires Théa
12, Rue Louis Blériot - Zone Industrielle du Brézet
63017 CLERMONT-FERRAND – FRANÇA
Fabricante:
BENAC
10, Avenue Paul Langevin
17180 PÉRIGNY – FRANÇA
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
| Alemanha | Mydriasert |
| Áustria | Mydriasert |
| Bélgica | Mydriasert |
| Dinamarca | Mydriasert |
| Espanha | Mydriasert |
| Finlândia | Mydriasert |
| França | Mydriasert |
| Holanda | Mydriasert |
| Itália | Mydriasert |
| Luxemburgo | Mydriasert |
| Portugal | Mydriasert |
|
Reino Unido
|
Mydriasert |
| Suécia | Mydriasert |
Este folheto informativo foi revisto pela última vez em 11/2021
_______________________________________________________________
A informação que se segue destina-se apenas aos médicos e aos profissionais dos cuidados de saúde.
Não ingerir.
Antes de usar o medicamento, verifique a integridade da saqueta. Em caso de deterioração do invólucro da saqueta, a esterilidade deixa de estar assegurada; neste caso, use outro inserto oftálmico retirado de uma embalagem intacta.
POSOLOGIA
O profissional de saúde coloca um inserto oftálmico no saco conjuntival inferior do olho em questão, num máximo de duas horas antes da intervenção cirúrgica ou da investigação.
População pediátrica
Mydriasert está contraindicado em crianças com idade inferior a 12 anos.
Não existem dados quanto à utilização em crianças com idades entre os 12 e os 18 anos, pelo que Mydriasert não é recomendado para estes doentes.
MODO DE ADMINISTRAÇÃO
Cortar o bordo do invólucro ao longo da linha ponteada, abrir a saqueta e localizar o inserto. Puxar para baixo a pálpebra inferior, segurando-a entre o polegar e o indicador (A) e aplicar o inserto oftálmico no saco conjuntival inferior, usando a pinça esterilizada descartável fornecida na embalagem, a qual deve ser eliminada imediatamente após a utilização (B).
INSTRUÇÕES DE UTILIZAÇÃO
O inserto oftálmico não deve ser deixado mais de duas horas dentro do saco conjuntival inferior. O médico pode retirar o inserto oftálmico logo que a midríase seja considerada suficiente para a intervenção ou o procedimento a realizar; no máximo, o inserto deve ser retirado 30 minutos após dilatação suficiente da pupila. Em caso de desconforto, verificar se o inserto foi corretamente inserido na base do saco conjuntival inferior.
ADVERTÊNCIA: RETIRADA DO INSERTO OFTÁLMICO
Antes de uma intervenção ou procedimento, e logo que se obtenha a midríase desejada, o inserto oftálmico deve ser retirado do saco conjuntival inferior usando uma pinça cirúrgica esterilizada ou uma mecha esterilizada ou uma solução de lavagem ou irrigação esterilizada, puxando para baixo a pálpebra inferior (C).


Cirurgia
Aprokam

Folheto informativo: Informação para o utilizador
APROKAM 50 mg pó para solução injetável
Cefuroxima
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
1. O que é APROKAM e para que é utilizado
2. O que precisa de saber antes de lhe ser administrado APROKAM
3. Como é administrado APROKAM
4. Efeitos secundários possíveis
5. Como conservar APROKAM
6. Conteúdo da embalagem e outras informações
1. O que é APROKAM e para que é utilizado
• APROKAM contém uma substância ativa, cefuroxima (sob a forma de cefuroxima sódica), que pertence a um grupo de antibióticos chamados cefalosporinas. Os antibióticos são usados para matar as bactérias ou “germes” que causam infeções.
• Este medicamento será utilizado se for submetido a uma cirurgia aos olhos por ter cataratas (enevoamento do cristalino).
• O seu cirurgião oftalmológico administrará este medicamento através de uma injeção no olho no final da cirurgia da catarata para prevenir uma infeção no olho.
2. O que precisa de saber antes de lhe ser administrado APROKAM
Não utilize APROKAM:
- se tem alergia (hipersensibilidade) à cefuroxima ou a qualquer outro antibiótico do tipo das cefalosporinas.
Advertências e precauções
Fale com o seu médico ou farmacêutico ou enfermeiro antes de utilizar APROKAM:
• se é alérgico a outros antibióticos como a penicilina,
• se está em risco de contrair uma infeção devido a uma bactéria chamada Staphylococcus aureus resistente à meticilina,
• se está em risco de contrair uma infeção grave,
• se lhe foi diagnosticada uma catarata complicada,
• se está planeada uma cirurgia ocular combinada,
• se tem uma doença grave da tiroide.
APROKAM é apenas administrado através de uma injeção no olho (injeção intracameral).
APROKAM deve ser administrado nas condições de assepsia (o que significa em condições de higiene e isentas de germes) da cirurgia da catarata.
Um frasco para injetáveis de APROKAM deve ser usado apenas para um doente.
Outros medicamentos e APROKAM
Informe o seu médico ou farmacêutico se estiver a tomar, tiver tomado recentemente ou se vier a tomar outros medicamentos.
Gravidez e amamentação
• Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de lhe ser administrado este medicamento.
• APROKAM ser-lhe-á apenas administrado se os benefícios compensarem os possíveis riscos.
APROKAM contém sódio
Este medicamento contém menos do que 1 mmol (23 mg) de sódio por dose ou seja, é praticamente “isento de sódio”.
3. Como é administrado APROKAM
• As injeções de APROKAM serão administradas por um cirurgião oftalmológico no final da cirurgia da catarata.
• APROKAM é fornecido sob a forma de um pó estéril que, antes de ser administrado, é dissolvido numa solução de soro fisiológico para injetáveis.
Se lhe for administrado menos ou mais APROKAM do que deveria
A sua medicação ser-lhe-á geralmente administrada por um profissional de saúde. Se pensa que se podem ter esquecido de lhe administrar uma dose ou que recebeu uma dose do medicamento superior à que deveria, por favor informe o seu médico ou enfermeiro.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico ou enfermeiro.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
O seguinte efeito secundário é muito raro (pode afetar até 1 em 10.000 pessoas):
• Reação alérgica grave que provoca dificuldade em respirar ou tonturas.
O seguinte efeito secundário é reportado com uma frequência “Desconhecida” (não pode ser calculada a partir dos dados disponíveis):
• Edema macular (visão turva ou distorcida na proximidade ou no centro do seu campo de visão).
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar APROKAM
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize APROKAM após o prazo de validade impresso no rótulo do frasco para injetáveis e na embalagem exterior após EXP. O prazo de validade corresponde ao último dia do mês indicado.
Conservar abaixo de 25ºC. Manter o frasco para injetáveis na embalagem exterior para proteger da luz.
Apenas para uma única utilização.
Após reconstituição: o produto deve ser usado imediatamente.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de APROKAM
- A substância ativa é a cefuroxima (sob a forma de cefuroxima sódica).
- Cada frasco para injetáveis contém 50 mg de cefuroxima.
- Após reconstituição, 0,1 ml de solução contêm 1 mg de cefuroxima.
- Não contém outros componentes.
Qual o aspeto de APROKAM e conteúdo da embalagem
APROKAM é um pó para solução injetável branco ou esbranquiçado, fornecido num frasco para injetáveis de vidro.
Cada embalagem contém um ou dez ou vinte frascos para injetáveis ou 10 frascos para injetáveis com 10 agulhas estéreis com filtro. É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
LABORATOIRES THEA
12 Rue Louis Blériot
63017 CLERMONT-FERRAND Cedex 2
França
Fabricante
BIOPHARMA S.R.L.
Via Delle Gerbere, 22/30 (loc. S. PALOMBA)
00134 ROMA (RM)
Itália
ou
LABORATOIRES THEA
12 Rue Louis Blériot
63017 CLERMONT-FERRAND Cedex 2
França
Este medicamento encontra-se autorizado nos seguintes Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Alemanha, Áustria, Bélgica, Bulgária, Dinamarca, Eslováquia, Eslovénia, Finlândia, França, Holanda, Itália, Islândia, Luxemburgo, Noruega, Polónia, Portugal, República Checa, Roménia, Suécia, Reino Unido: APROKAM
Chipre, Grécia, Espanha: PROKAM
Irlanda: APROK
Este folheto foi revisto pela última vez em 08/2019
A informação que se segue destina-se apenas aos profissionais de saúde:
Incompatibilidades
Não se encontra descrita na literatura qualquer incompatibilidade com os produtos mais frequentemente utilizados na cirurgia da catarata. Este medicamento não deve ser misturado com outros medicamentos, exceto os que se encontram mencionados em seguida [solução injetável de cloreto de sódio a 9 mg/ml (0,9%)].
Como preparar e administrar APROKAM
Frasco para injetáveis para uma única utilização apenas para administração intracameral.
APROKAM deve ser administrado após reconstituição através de uma injeção intraocular na câmara anterior do olho (injeção intracameral) efetuada por um cirurgião oftalmológico, nas condições de assepsia recomendadas para a cirurgia da catarata.
A solução reconstituída deve ser visualmente inspecionada e só deverá ser utilizada se a solução for incolor ou ligeiramente amarelada e isenta de partículas visíveis.
O produto deve ser usado imediatamente após a reconstituição e não deve ser reutilizado.
A dose recomendada para a cefuroxima é de 1 mg em 0,1 ml de solução injetável de cloreto de sódio a 9 mg/ml (0,9%).
NÃO INJETE UMA DOSE SUPERIOR À RECOMENDADA.
O frasco para injetáveis destina-se apenas a uma única utilização.
Um frasco para injetáveis apenas para um doente. Cole a etiqueta destacável do frasco para injetáveis na ficha do doente.
Siga as seguintes instruções para preparar o APROKAM para administração intracameral:
1. Verifique a integridade da cápsula de abertura fácil antes de a retirar.
2. Desinfete a superfície da tampa de borracha antes de efetuar o passo 3.
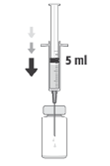
3. Insira a agulha na vertical no centro da tampa do frasco para injetáveis, mantendo o frasco para injetáveis na posição vertical. Em seguida, usando uma técnica asséptica injete no frasco para injetáveis 5 ml de uma solução injetável de cloreto de sódio a 9 mg/ml (0,9%).
4. Agite suavemente até que a solução esteja isenta de partículas visíveis.
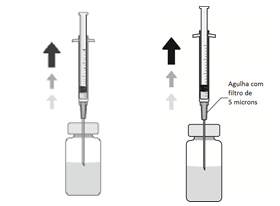
5. Adapte uma agulha estéril (18G x 1½”, 1,2 mm x 40 mm) com um filtro de 5 microns (membrana de copolímero acrílico num suporte de nylon não tecido) numa seringa estéril de 1 ml (a agulha estéril com um filtro de 5 microns poderá ser fornecida na embalagem). Empurre esta seringa verticalmente no centro da tampa do frasco para injetáveis, mantendo o frasco para injetáveis na posição vertical.
6. Aspire assepticamente, pelo menos 0,1 ml de solução.
7. Retire a agulha da seringa e adapte na seringa uma cânula apropriada para a câmara anterior.

8. Cuidadosamente deve expelir o ar da seringa e ajustar a dose para atingir a marca correspondente a 0,1 ml na seringa. A seringa encontra-se preparada para a injeção.
Após a utilização, rejeite a solução reconstituída que ainda restar. Não a guarde para uma utilização subsequente.
Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com as exigências locais. Elimine as agulhas utilizadas nos contentores adequados.

Cirurgia
Mydrane

Folheto informativo: Informação para o doente
MYDRANE 0,2 mg/ml + 3,1 mg/ml + 10 mg/ml solução injetável
Tropicamida / cloridrato de fenilefrina / cloridrato de lidocaína mono-hidratado
Leia com atenção todo este folheto antes de lhe ser administrado este medicamento, pois contém informação importante para si.
Conserve este folheto. Pode ter necessidade de o ler novamente.
Caso ainda tenha dúvidas, fale com o seu médico, farmacêutico ou enfermeiro.
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
1. O que é MYDRANE e para que é utilizado
2. O que precisa de saber antes de lhe ser administrado MYDRANE
3. Como é administrado MYDRANE
4. Efeitos secundários possíveis
5. Como conservar MYDRANE
6. Conteúdo da embalagem e outras informações
1. O que é MYDRANE e para que é utilizado
O que é MYDRANE
Este medicamento é uma solução, a qual lhe vai ser injetada no olho.
Contém 3 substâncias ativas:
• tropicamida que pertence a um grupo de medicamentos que bloqueia a passagem de impulsos por determinados nervos (conhecido como anticolinérgico),
• fenilefrina (sob a forma de cloridrato de fenilefrina) que pertence a um grupo de medicamentos que mimetiza os efeitos dos impulsos transmitidos por determinados nervos (o qual pertence aos simpaticomiméticos alfa),
• lidocaína (sob a forma de cloridrato de lidocaína mono-hidratado) que pertence a uma classe de medicamentos denominada anestésicos locais do tipo amida.
Para que é utilizado
Este medicamento é utilizado apenas em adultos.
Ser-lhe-á administrado pelo seu cirurgião oftalmológico através de uma injeção no seu olho no início da cirurgia da catarata (enevoamento do cristalino), para dilatar a pupila do seu olho (midríase) e anestesiar o seu olho durante o procedimento cirúrgico.
2. O que precisa de saber antes de lhe ser administrado MYDRANE
Não lhe deve ser administrado MYDRANE:
• se tem alergia à tropicamida, ao cloridrato de fenilefrina e/ou ao cloridrato de lidocaína mono-hidratado ou a qualquer outro componente deste medicamento (indicados na secção 6),
• se tem alergia aos anestésicos do tipo amida (articaína, bupivacaína, mepivacaína, prilocaína, ropivacaína),
• se tem alergia aos derivados da atropina.
Advertências e precauções
MYDRANE não é recomendado:
• na cirurgia da catarata quando combinada com um certo tipo de cirurgia ocular (vitrectomia),
• se a parte anterior (câmara anterior) do seu olho está inchada,
• se tem antecedentes de pressão ocular elevada aguda (glaucoma de angulo estreito agudo).
Deve falar com o seu médico especialmente se tiver:
• pressão arterial elevada (hipertensão),
• espessamento da parede arterial (aterosclerose),
• qualquer doença no coração e em particular se esta afetar o ritmo cardíaco,
• uma contraindicação para medicamentos que aumentem a pressão arterial (aminas vasopressoras: adrenalina, noradrenalina, dopamina, dobutamina) por via oral,
• hiperatividade da glândula tiroide (hipertiroidismo),
• perturbações na glândula da próstata,
• crises de epilepsia,
• qualquer doença no fígado ou problemas nos rins,
• qualquer problema com a sua respiração,
• perda da função muscular e fraqueza (miastenia gravis).
Outros medicamentos e MYDRANE
Informe o seu médico ou farmacêutico se estiver a tomar, tiver tomado recentemente ou se vier a tomar outros medicamentos.
Gravidez, amamentação e fertilidade
Este medicamento não deve ser utilizado:
• durante a gravidez,
• durante a amamentação.
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento.
Condução de veículos e utilização de máquinas
MYDRANE tem uma influência moderada na capacidade de conduzir e utilizar máquinas. Consequentemente, não deve conduzir e/ou utilizar máquinas até que a sua visão volte ao normal.
MYDRANE contém sódio
Este medicamento contém menos do que 1 mmol (23 mg) de sódio por unidade de dose, ou seja, é praticamente “isento de sódio”.
3. Como é administrado MYDRANE
Apenas lhe será administrado este medicamento caso já tenha demonstrado, numa avaliação pré-operatória, uma dilatação satisfatória da pupila com uma terapêutica midriática tópica.
Dose e modo de administração
• MYDRANE injetável ser-lhe-á administrado por um cirurgião oftalmológico, sob anestesia local, no início da cirurgia da catarata.
• A dose recomendada é de 0,2 ml de solução, numa injeção única. Não deverá ser administrada uma dose adicional, pois não foi demonstrado efeito adicional e além disso foi observado um aumento da perda de células endoteliais (células da camada que reveste a superfície posterior da córnea).
• É utilizada a mesma dose para adultos e idosos.
Se lhe for administrado mais, ou menos, MYDRANE do que deveria
O seu medicamento ser-lhe-á administrado por um cirurgião oftalmológico. É pouco provável que lhe seja administrada uma dose excessiva.
A sobredosagem pode causar um aumento na perda de células endoteliais (células da camada que cobre a superfície posterior da córnea).
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico ou enfermeiro.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Complicações mais graves e bem conhecidas que ocorrem durante ou após a cirurgia da catarata:
Pouco frequentes: podem afetar até 1 em 100 pessoas
• Lesão do cristalino (rutura da cápsula posterior),
• Inchaço da retina (edema macular cistoide).
Nesta situação procure aconselhamento médico urgente.
Outros efeitos secundários:
Pouco frequentes: podem afetar até 1 em 100 pessoas
• Dores de cabeça,
• Inchaço da córnea (queratite), pressão no olho aumentada, vermelhidão do olho (hiperemia ocular),
• Pressão arterial elevada (hipertensão).
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar MYDRANE
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na cartonagem, blister e ampola. O prazo de validade corresponde ao último dia do mês indicado.
O medicamento não necessita de quaisquer precauções especiais de conservação.
Apenas para uma utilização única. Este medicamento deve ser utilizado imediatamente após a primeira abertura da ampola.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de MYDRANE
• As substâncias ativas são: tropicamida 0,04 mg, cloridrato de fenilefrina 0,62 mg e cloridrato de lidocaína mono-hidratado 2 mg por cada dose de 0,2 ml, o que corresponde a 0,2 mg de tropicamida, 3,1 mg de cloridrato de fenilefrina e 10 mg de cloridrato de lidocaína mono-hidratado por cada 1 ml.
• Os outros componentes são: cloreto de sódio, fosfato disódico dodeca-hidratado, fosfato disódico di-hidratado, edetato dissódico, água para preparações injetáveis.
Qual o aspeto de MYDRANE e conteúdo da embalagem
MYDRANE é uma solução injetável límpida, ligeiramente amarela-acastanhada, praticamente isenta de partículas visíveis, acondicionada numa ampola de vidro castanho de 1 ml. Cada ampola estéril contém 0,6 ml de solução injetável e encontra-se acondicionada separadamente ou em conjunto com uma agulha estéril com filtro de 5 micrómetros num blister selado papel/PVC.
Cada embalagem contém 1 ou 20 ou 100 ampolas estéreis com agulha(s) estéril(eis) com filtro de 5 micrómetros fornecidas separadamente ou em conjunto no mesmo blister. As agulhas com filtro de 5 micrómetros devem ser utilizadas apenas para retirar o conteúdo da ampola. Todos os componentes são apenas para uma única utilização.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires THEA
12, Rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
Fabricante
Delpharm Tours
Rue Paul Langevin
37170 Chambray les tours
França
ou
Laboratoires Thea
12, Rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
Este medicamento encontra-se autorizado nos seguintes Estados membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Alemanha, Áustria, Bélgica, Bulgária, Chipre, Croácia, Dinamarca, Eslováquia, Eslovénia, Finlândia, França, Grécia, Holanda, Itália, Irlanda, Luxemburgo, Polónia, Portugal, Roménia, Reino Unido, República Checa, Suécia Mydrane
Espanha, Irlanda Fydrane
Noruega Mydane
Este folheto foi revisto pela última vez em 09/2023
A informação que se segue destina-se apenas aos profissionais de saúde:
Incompatibilidades
Não foram reportadas na literatura nem durante os ensaios clínicos, incompatibilidades entre os produtos mais frequentemente utilizados na cirurgia da catarata e as substâncias ativas. Para os viscoelásticos comuns, tal foi também confirmado pelos de testes de interação farmacêutica.
Advertência
Não utilizar se o blister estiver danificado ou rasgado. Abrir apenas sob condições asséticas. Garante-se que o conteúdo do blister se encontra estéril.
Como preparar e administrar MYDRANE
Solução para utilização apenas num olho por via intracameral.
MYDRANE deve ser administrado por injeção intraocular na câmara anterior do olho (injeção intracameral), por um cirurgião oftalmológico, nas condições de assepsia recomendadas para a cirurgia da catarata.
Antes da injeção intracameral, a solução deve ser visualmente inspecionada e só deverá ser utilizada se estiver límpida ligeiramente amarela-acastanhada e praticamente isenta de partículas visíveis.
A dose recomendada é de 0,2 ml de MYDRANE; não deverá ser administrada uma dose adicional, pois não foi demonstrado efeito adicional e foi observado um aumento da perda de células endoteliais.
O produto deve ser utilizado imediatamente após a primeira abertura da ampola e não deve ser reutilizado em outro olho ou em qualquer outro doente.
Apenas para a apresentação em kit (i.e. blister contendo uma ampola e uma agulha): cole a etiqueta do blister no processo do doente.
Siga as seguintes instruções para preparar MYDRANE para a injeção intracameral:
1. Verifique o blister antes de o abrir para garantir que está intacto. Retire a película do blister em condições asséticas para garantir a esterilidade do conteúdo.

2. Abra a ampola estéril que contém o produto. A ampola deve ser aberta pelo ponto de quebra (OPC) da seguinte forma: segure a parte de baixo da ampola com o polegar a apontar para o ponto colorido. Segure o topo da ampola com a outra mão, posicionando o polegar no ponto colorido e pressione para trás de forma a quebrar no ponto de quebra que está abaixo do ponto colorido.
3. Encaixe a agulha estéril com filtro de 5 microns (fornecida) numa seringa estéril. Retire a proteção da agulha estéril com filtro de 5 microns e aspire, pelo menos, 0,2 ml de solução injetável da ampola para a seringa.
4. Retire a agulha da seringa e adapte na seringa uma cânula apropriada para a câmara anterior.
5. Cuidadosamente deve expelir o ar da seringa. Ajuste até aos 0,2 ml. A seringa encontra-se pronta para ser utilizada.
6. Injete lentamente os 0,2 ml contidos na seringa na câmara anterior do olho, numa injeção única, através da paracentese ou da incisão principal.
Após a utilização, rejeite a solução que ainda reste. Não a guarde para uma utilização subsequente.
Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com as exigências locais. Elimine as agulhas utilizadas nos contentores adequados.
- Glaucoma

Glaucoma
Timabak

Folheto informativo: Informação para o utilizador
Timabak 5 mg/ml colírio, solução
Maleato de timolol
Leia com atenção todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico ou farmacêutico. Ver secção 4.
O que contém este folheto:
- O que é Timabak e para que é utilizado
- O que precisa de saber antes de utilizar Timabak
- Como utilizar Timabak
- Efeitos indesejáveis possíveis
- Como conservar Timabak
- Conteúdo da embalagem e outras informações
1. O que é Timabak e para que é utilizado
Timabak, 5 mg/ml, colírio, solução contém maleato de timolol, um bloqueador beta para uso oftálmico.
Timabak, 5 mg/ml, colírio, solução está indicado no tratamento de certas afeções do olho que envolvem aumento da pressão intraocular (glaucoma, hipertensão intraocular)
2. O que precisa de saber antes de utilizar Timabak
Não utilize Timabak
- se tem alergia (hipersensibilidade) ao maleato de timolol, bloqueadores beta ou a qualquer outro componente deste medicamento (indicados na secção 6);
- se tem ou já teve problemas respiratórios tais como asma, bronquite obstrutiva crónica grave (doença pulmonar grave que possa causar pieira, dificuldade em respirar e/ou tosse prolongada);
- se tem um ritmo cardíaco lento, insuficiência cardíaca ou alterações do ritmo cardíaco (batimento cardíaco irregular);
- se sofre de feocromocitoma não tratado;
- se sofre de hipotensão;
- em caso de tratamento pela floctafenina; Este medicamento não deve em geral, ser associado à amiodarona.
Advertências e precauções
Fale com o seu médico ou farmacêutico antes de utilizar Timabak.
Não injetar, não ingerir.
O tratamento com Timabak, 5 mg/ml, colírio, solução necessita de controlo oftalmológico regular.
Não interromper o tratamento, sem recomendação do seu médico.
Evitar o uso de lentes de contacto durante o tratamento.
Antes de utilizar este medicamento, informe o seu médico se tem agora ou se já teve no passado:
- doença cardíaca coronária (os sintomas podem incluir dor torácica ou aperto no peito, falta de ar ou sufoco), insuficiência cardíaca, pressão arterial baixa,
- alterações do ritmo cardíaco tais como ritmo cardíaco lento,
- problemas respiratórios, asma ou doença pulmonar obstrutiva crónica,
- problemas de circulação sanguínea (como doença ou síndrome de Raynaud),
- diabetes, uma vez que o maleato de timolol pode mascarar os sinais e sintomas de nível baixo de açúcar no sangue,
- hiperatividade da glândula tiroide, uma vez que o maleato de timolol pode mascarar sinais e sintomas.
Foram descritos casos de descolamento coroideu após cirurgia de filtração.
Antes de uma cirurgia informe o seu médico que está a utilizar Timabak, pois o maleato de timolol pode alterar o efeito de alguns medicamentos utilizados durante a anestesia.
Crianças e adolescentes
Os colírios, solução contendo timolol devem, de uma maneira geral, ser utilizados com precaução em doentes mais jovens.
Em recém-nascidos, bebés e crianças o timolol deve ser utilizado com extrema precaução.
Caso ocorra tosse, respiração ruidosa, alterações na respiração ou pausas estranhas na respiração (apneia), a utilização do medicamento deve ser imediatamente interrompida.
Contacte o seu médico o mais rapidamente possível.
Poderá ser útil ter um monitor portátil de apneia.
Timabak foi estudado em bebés e crianças com idades compreendidas entre os 12 dias e os 5 anos, nos quais houve um aumento da pressão no(s) olho(s) ou aos quais foi diagnosticado glaucoma.
Para mais informações, fale com o seu médico.
Outros medicamentos e Timabak
Informe o seu médico ou farmacêutico se estiver a utilizar, tiver utilizado recentemente ou se vier a utilizar outros medicamentos.
Se o seu médico lhe prescrever um outro colírio, deverá instilar esse colírio 15 minutos antes de Timabak, 5 mg/ml, colírio, solução.
O Timabak pode afetar ou ser afetado por outros medicamentos que esteja a tomar, incluindo outros colírios para o tratamento do glaucoma.
Informe o seu médico se estiver a tomar ou pretender tomar medicamentos para baixar a pressão arterial, medicamentos para o coração ou medicamentos para tratar a diabetes, quinidina (utilizada no tratamento de doenças do coração e alguns tipos de malária) e antidepressivos como a fluoxetina e a paroxetina.
Uma vigilância oftalmológica é necessária em caso de tratamento concomitante com um colírio contendo adrenalina (risco de midríase).
Evitar o uso de amiodarona durante o tratamento com Timabak, 5 mg/ml, colírio, solução dado o risco de ocorrerem perturbações do automatismo e da condução cardíaca.
Informe o seu médico se estiver a tomar floctafenina, amiodarona.
Gravidez e amamentação
Não utilize Timabak se está grávida a não ser que o seu médico considere necessário.
Não utilize Timabak se está a amamentar. O maleato de timolol pode passar para o leite.
Consulte o seu médico para obter aconselhamento sobre a toma de medicamentos durante a amamentação.
Condução de veículos e utilização de máquinas
Durante a condução de veículos ou a utilização de máquinas deve ter em consideração que ocasionalmente podem ocorrer perturbações visuais incluíndo alterações na visão, visão dupla, pálpebra descaída, episódios frequentes de visão turva ligeira e temporária e ocasionalmente episódios de tonturas ou fadiga.
Excipientes
Este medicamento contém 0,361 mg de fosfatos em cada gota que é equivalente a 11,28 mg/ml.
Se sofrer de lesão grave na camada transparente na parte da frente do olho (córnea), os fosfatos podem causar em casos muito raros zonas nubladas na córnea devido à acumulação de cálcio durante o tratamento.
3. Como utilizar Timabak
Utilize este medicamento exatamente como indicado pelo seu médico ou farmacêutico.
Fale com o seu médico ou farmacêutico se tiver dúvidas.
A posologia habitual é de 1 gota de colírio no olho afetado, 2 vezes por dia.
Este medicamento destina-se a uma aplicação tópica para uso oftálmico.
Lavar cuidadosamente as mãos antes da instilação.
Instilar uma gota do colírio no fundo do saco conjuntival do olho afetado, olhando para cima e puxando ligeiramente a pálpebra para baixo, evitando o contacto da extremidade do frasco com o olho ou com as pálpebras.
Depois de utilizar Timabak, pressione com um dedo no canto do olho, junto ao nariz durante 2 minutos.
Isto impede que o maleato de timolol vá para outras partes do corpo.
Colocar a tampa no frasco após a utilização.
O tempo de aparecimento de cada gota é mais longo do que num frasco de colírio clássico.
O medicamento deve ser aplicado em geral de manhã e à noite (2 instilações diárias).
Não interromper o tratamento a não ser por recomendação médica.
Utilização em crianças
Posologia
Antes da utilização de timolol deve ser realizado um exame médico cuidadoso.
O seu médico irá avaliar cuidadosamente os riscos e os benefícios antes de considerar o tratamento com timolol.
Caso os benefícios sejam superiores aos riscos, é recomendada a utilização da dosagem mais baixa disponível uma vez ao dia.
Tendo em consideração a "utilização em crianças", a concentração de 1 mg/ml poderá ser suficiente para controlar a pressão do olho.
Caso a pressão não seja controlada de forma adequada com esta dosagem, poderá ser necessária a aplicação duas vezes ao dia com um intervalo de 12 horas entre as aplicações.
Os doentes, especialmente os recém-nascidos, devem ser cuidadosamente observados durante uma a duas horas após a primeira administração e devem ser cuidadosamente monitorizados para detetar o aparecimento de efeitos adversos até à realização da cirurgia.
Modo de administração
Deve ser instilada apenas uma gota de timolol em cada administração.
Após a instilação mantenha os olhos fechados durante 2 minutos e aplique pressão no canto do olho que está mais perto do nariz para prevenir que a gota de timolol colírio se espalhe pelo corpo.
Duração do tratamento
Nas crianças, o Timabak deverá ser prescrito como tratamento temporário.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos indesejáveis possíveis
Como todos os medicamentos, este medicamento pode causar efeitos indesejáveis, embora estes não se manifestem em todas as pessoas.
Pode continuar a aplicar as gotas, exceto se os efeitos forem graves.
Se estiver preocupado, fale com o seu médico ou farmacêutico.
Não pare de utilizar as gotas sem falar com o seu médico.
Como outros medicamentos de aplicação nos olhos, o maleato de timolol é absorvido pelo sangue. Isto pode causar efeitos indesejáveis semelhantes aos observados com medicamentos bloqueadores beta intravenosos e/ou orais, conforme aplicável.
A incidência de efeitos adversos após administração oftálmica é inferior a quando os medicamentos são tomados, por exemplo, pela boca ou são injetados.
A lista de efeitos adversos inclui reações observadas dentro da classe dos bloqueadores beta quando estes são utilizados para tratar afeções dos olhos:
- Reações alérgicas generalizadas incluindo inchaço da pele (que pode ocorrer em áreas como a face e os membros e pode obstruir as vias aéreas provocando dificuldade em respirar e engolir), urticária (ou erupção cutânea que provoca comichão), erupção cutânea localizada e generalizada, comichão, dermatite exfoliativa, reação alérgica aguda grave com risco de vida. Pode contribuir para a sensibilidade/refratariedade (resistência à adrenalina) ao tratamento da reação anafilática/hipersensibilidade. Lúpus eritematoso sistémico.
- Níveis baixos ou níveis elevados de açúcar no sangue.
- Dificuldade em dormir (insónia), depressão, pesadelos, perda de memória, alucinação.
- Desmaio, acidente vascular cerebral, diminuição do fornecimento de sangue ao cérebro, aumento dos sinais e sintomas de miastenia grave (doença muscular), dores nas articulações, tonturas, sensações fora do normal (como formigueiro), dor de cabeça.
- Sinais e sintomas de irritação ocular (por exemplo, ardor, picadas, comichão, aumento do lacrimejo, vermelhidão), conjuntivite, inflamação da pálpebra, inflamação da córnea, visão turva e descolamento da camada abaixo da retina que contém vasos sanguíneos, após cirurgia de filtração, o que pode causar distúrbios visuais, sensibilidade da córnea diminuída, olhos secos, erosão da córnea (danos na camada da frente do globo ocular), pálpebra superior descaída (ficando o olho meio fechado), visão dupla, perturbações visuais incluindo alterações na refração (devidas por vezes à suspensão do tratamento com o colírio miótico).
- Ritmo cardíaco lento, dor no peito, palpitações, edema (acumulação de líquidos), alterações no ritmo ou na velocidade dos batimentos cardíacos, insuficiência cardíaca congestiva (doença do coração com falta de ar e inchaço dos pés e pernas devido à acumulação de fluidos), um tipo de distúrbio do ritmo cardíaco, ataque cardíaco, insuficiência cardíaca, agravamento da angina de peito e da insuficiência arterial, vasodilatação.
- Pressão arterial baixa, fenómeno de Raynaud, mãos e pés frios, cãibras nas pernas e/ou dores nas pernas durante o caminhar (claudicação).
- Sensação de aperto das vias respiratórias nos pulmões (predominantemente em doentes com doença pré-existente), edema pulmonar, dificuldade em respirar, insuficiência respiratória, tosse.
- Distúrbios do paladar, náuseas, azia, indigestão, diarreia, boca seca, dor abdominal, vómitos.
- Queda de cabelo, erupções na pele com aspeto branco prateado (erupção cutânea psoriasiforme) ou agravamento da psoríase, erupções cutâneas.
- Dor muscular não causada por exercício físico, dor articular.
- Disfunção sexual, diminuição da libido, impotência, doença de Peyronie.
- Fraqueza muscular /cansaço.
Se sofrer de lesão grave na camada transparente na parte da frente do olho (córnea), os fosfatos podem causar em casos muito raros zonas nubladas na córnea devido à acumulação de cálcio durante o tratamento.
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico ou farmacêutico. Também poderá comunicar efeitos indesejáveis diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos indesejáveis, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
(preferencialmente)
ou através dos seguintes contactos: Direção de Gestão do Risco de Medicamentos Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita) E-mail: farmacovigilancia@infarmed.pt
5. Como conservar Timabak
Não conservar acima de 25ºC.
Uma vez aberto, o frasco não deve ser conservado para além de 8 semanas.
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso no frasco e na embalagem, após VAL.O prazo de validade corresponde ao último dia do mês indicado.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico.
Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza.
Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Timabak
- A substância ativa é o maleato de timolol, correspondente a 5 mg de timolol. Cada ml de colírio, solução contém 5 mg de timolol.
- Os outros componentes são o hidrogenofosfato de sódio dodecahidratado, fosfato monossódico di-hidratado e água para preparações injetáveis.
Qual o aspeto de Timabak e conteúdo da embalagem
Timabak apresenta-se em frascos conta-gotas com 5 ml e 10 ml, equipados com um dispositivo que protege o colírio contra a contaminação microbiana durante o período de utilização.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires Théa
12 Rue Louis Blériot
Zone Industrielle du Brezet
63017 Clermont-Ferrand
FRANÇA
Fabricante
Excelvision (Fab. Annonay)
Rue de la Lombardière
F-07100 Annonay
Farmila - Thea Farmaceutici S.p.A. (Fab. Settimo Milanese)
Via Enrico Fermi, 50
I-20019 Settimo Milanese - Milano
Este folheto foi revisto pela última vez em 06/2025

Glaucoma
Timogel

Folheto informativo: informação para o doente
TIMOGEL 1 mg/g, gel oftálmico em recipiente unidose
Timolol
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico, farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Ver secção 4.
Neste folheto:
1. O que é TIMOGEL e para que é utilizado
2. O que precisa de saber antes de utilizar TIMOGEL
3. Como utilizar TIMOGEL
4. Efeitos secundários possíveis
5. Como conservar TIMOGEL
6. Conteúdo da embalagem e outras informações
1. O que é TIMOGEL e para que é utilizado
Este medicamento é um bloqueador beta administrado por via oftálmica.
É utilizado no tratamento de certos tipos de doenças oculares que envolvem hipertensão intraocular (glaucoma e hipertensão ocular).
2. O que precisa de saber antes de utilizar TIMOGEL
Não utilize TIMOGEL gel oftálmico em recipiente unidose
- se tem alergia ao maleato de timolol, aos bloqueadores beta ou a qualquer outro componente deste medicamento (indicados na secção 6),
- se tem ou já teve problemas respiratórias tais como, asma, bronquite obstrutiva crónica grave (doença pulmonar grave que pode causar pieira, dificuldade em respirar e/ou tosse prolongada),
- se tem um ritmo cardíaco lento, insuficiência cardíaca ou alterações do ritmo cardíaco (batimento cardíaco irregular),
- em caso de feocromocitoma não tratado (produção excessiva de uma hormona que provoca hipertensão arterial grave),
- em caso de distrofias da córnea (doenças degenerativas da córnea).
Advertências e precauções
Fale com o seu médico, farmacêutico ou enfermeiro antes de utilizar TIMOGEL.
Não interrompa subitamente o tratamento sem a indicação do seu médico.
Não injete, não ingira.
Se utiliza TIMOGEL, deverá efetuar controlos regulares da pressão intraocular e da córnea.
Antes de utilizar este medicamento, informe o seu médico se tem agora ou se já teve no passado:
- doença cardíaca coronária (os sintomas podem incluir dor torácica ou aperto no peito, falta de ar ou sufoco), insuficiência cardíaca, pressão arterial baixa,
- alterações do ritmo cardíaco, tais como ritmo cardíaco lento,
- problemas respiratórios, asma ou doença pulmonar obstrutiva crónica (doença pulmonar grave que pode causar pieira, dificuldade em respirar e/ou tosse prolongada),
- problemas de circulação sanguínea (como doença de Raynaud ou síndrome de Raynaud),
- diabetes, uma vez que o maleato de timolol pode mascarar os sinais e os sintomas de um nível baixo de açúcar no sangue,
- hiperatividade da glândula tiroide, uma vez que o maleato de timolol pode mascarar os sinais e os sintomas,
- feocromocitoma tratado,
- psoríase,
- doença da córnea,
- doença metabólica.
Precauções relativas ao uso de lentes de contacto
Evite utilizar lentes de contacto durante o tratamento, pois verifica-se uma menor secreção de lágrimas; tal encontra-se geralmente associado aos bloqueadores beta.
Antes de uma cirurgia informe o seu médico que está a utilizar TIMOGEL, pois o maleato de timolol pode alterar o efeito de alguns medicamentos utilizados durante a anestesia.
Outros medicamentos e TIMOGEL
Se o seu médico lhe prescreveu outro tipo de colírio, deverá instilar esse colírio 15 minutos antes do TIMOGEL.
TIMOGEL pode afetar ou ser afetado por outros medicamentos que esteja a tomar, incluindo outros colírios para o tratamento do glaucoma.
Informe o seu médico se estiver a tomar ou pretender tomar medicamentos para baixar a pressão arterial, medicamentos para o coração ou medicamentos para tratar a diabetes, quinidina (utilizada no tratamento de doenças do coração e alguns tipos de malária) e antidepressivos como a fluoxetina e a paroxetina.
Informe o seu médico ou farmacêutico se estiver a tomar, tiver tomado recentemente, ou se vier a tomar outros medicamentos.
Gravidez, amamentação e fertilidade
Não utilize TIMOGEL se está grávida, exceto se o seu médico considerar necessária a sua utilização.
Não utilize TIMOGEL se está a amamentar. O maleato de timolol pode passar para o seu leite.
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento.
Condução de veículos e utilização de máquinas
Poderá sentir uma turvação temporária da visão após a administração oftálmica do medicamento. Espere até recuperar a visão normal antes de conduzir ou utilizar qualquer máquina. TIMOGEL pode também induzir outros efeitos secundários (tonturas, fadiga) que provavelmente afetarão a sua capacidade de conduzir ou utilizar máquinas. Se tem quaisquer dúvidas, por favor consulte o seu médico.
Desporto
A substância ativa deste medicamento pode originar resultados positivos nas análises para pesquisa de fármacos.
3. Como utilizar TIMOGEL
Utilize este medicamento exatamente como indicado pelo seu médico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
Posologia
- Adultos
A dose habitual é de uma gota no(s) olho(s) afetado(s), uma vez por dia, de manhã.
Cada recipiente unidose contém uma quantidade suficiente de gel para tratar ambos os olhos.
- Crianças e adolescentes
Não existe experiência em crianças e adolescentes. Consequentemente, não se recomenda a utilização deste gel oftálmico nestes doentes.
- Doentes idosos
Não é necessário qualquer ajuste posológico.
Frequência de administração
Aplique uma gota no(s) olho(s) afetado(s), uma vez por dia, de manhã.
Modo de administração
Este medicamento destina-se a ser administrado no olho (uso oftálmico).
Apenas para uso único.
- Lave cuidadosamente as mãos antes de utilizar o seu gel oftálmico.
- Vire o recipiente unidose para baixo e agite vigorosamente antes de aplicar a gota.
- Evite o contacto da extremidade do recipiente com o olho ou com as pálpebras.
- Puxe a pálpebra inferior do olho afetado suavemente para baixo, enquanto olha para cima e instile a gota no olho.
- Depois de utilizar TIMOGEL, pressione com um dedo no canto do olho, junto ao nariz durante 2 minutos. Isto impede que o maleato de timolol vá para outras partes do corpo.
- Após a utilização, rejeite o recipiente de dose unitária. Não o conserve para uma posterior utilização.
Se utilizar mais TIMOGEL do que deveria
Entre outros efeitos, poderá sentir como se tivesse tonturas, ou poderá sentir dificuldades respiratórias ou sentir que a sua frequência cardíaca diminuiu.
CONSULTE IMEDIATAMENTE O SEU MÉDICO OU FARMACÊUTICO
Caso se tenha esquecido de utilizar TIMOGEL
Não utilize uma dose a dobrar para compensar a dose que se esqueceu. Instile a dose de que se esqueceu logo que seja possível. Contudo, se estiver já perto da dose seguinte, não utilize a dose de que se esqueceu.
Se parar de utilizar TIMOGEL
A pressão no interior do seu olho pode aumentar e afetar a sua visão.
Não interrompa subitamente o tratamento sem a indicação do seu médico.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos secundários possíveis
Como todos os medicamentos, TIMOGEL gel oftálmico em recipiente unidose, pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Pode continuar a aplicar as gotas, exceto se os efeitos forem graves. Se estiver preocupado, fale com o seu médico ou farmacêutico. Não interrompa a utilização de TIMOGEL sem falar com o seu médico.
Como outros medicamentos de aplicação nos olhos, o maleato de timolol é absorvido pelo sangue. Isto pode causar efeitos secundários semelhantes aos observados com medicamentos bloqueadores beta intravenosos e/ou orais, conforme aplicável. A incidência de efeitos adversos após administração oftálmica é inferior a quando os medicamentos são tomados, por exemplo, pela boca ou são injetados. A lista de efeitos adversos inclui reações observadas dentro da classe dos bloqueadores beta quando estes são utilizados para tratar afeções dos olhos:
• Lúpus eritematoso disseminado, reações alérgicas generalizadas incluindo inchaço da pele (que pode ocorrer em zonas como a face e os membros e pode obstruir as vias aéreas provocando dificuldade em engolir ou respirar), urticária (ou erupção cutânea que provoca comichão), erupção cutânea localizada ou generalizada, comichão, reação alérgica aguda grave com risco de vida.
• Níveis baixos de glucose no sangue.
• Dificuldade em dormir (insónia), depressão, pesadelos, perda de memória, alucinação.
• Desmaio, acidente vascular cerebral, diminuição do fornecimento de sangue ao cérebro, aumento dos sinais e sintomas de miastenia grave (doença muscular), tonturas, sensações fora do normal (como formigueiro), dor de cabeça.
• Sinais e sintomas de irritação ocular (por exemplo, ardor, picadas, comichão, aumento do lacrimejo, vermelhidão), vermelhidão da conjuntiva, conjuntivite, inflamação da pálpebra, inflamação na córnea, visão turva, e descolamento da camada abaixo da retina que contém vasos sanguíneos, após cirurgia de filtração, o que pode causar perturbações visuais, sensibilidade da córnea diminuída, olhos secos, erosão da córnea, (lesão na camada da frente do globo ocular), pálpebra superior descaída (ficando o olho meio fechado), visão dupla, alterações na refração (devidas por vezes à suspensão do tratamento com o colírio miótico).
• Ritmo cardíaco lento, dor no peito, palpitações, edema (acumulação de líquidos), alterações no ritmo ou na velocidade dos batimentos cardíacos, insuficiência cardíaca congestiva (doença do coração com falta de ar e inchaço dos pés e pernas devido à acumulação de líquidos), um tipo de perturbação do ritmo cardíaco, ataque cardíaco, insuficiência cardíaca, cãibras nas pernas e/ou dores nas pernas ao caminhar (claudicação).
• Pressão arterial baixa, fenómeno de Raynaud, mãos e pés frios.
• Sensação de aperto das vias respiratórias nos pulmões (principalmente em caso de doença pré-existente), dificuldade em respirar, tosse.
• Perturbações do paladar, náuseas, indigestão, diarreia, boca seca, dor abdominal, vómitos.
• Queda de cabelo, erupções na pele com aspeto branco prateado (erupção cutânea psoriasiforme) ou agravamento da psoríase, erupções cutâneas.
• Dor muscular não causada por exercício físico.
• Disfunção sexual, diminuição da líbido, impotência.
• Fraqueza muscular/cansaço.
• Resultados positivos para os anticorpos antinucleares.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Fax: +351 21 798 73 97
Sítio da internet: http://extranet.infarmed.pt/page.seram.frontoffice.seramhomepage
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar TIMOGEL
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na cartonagem, na saqueta e no recipiente unidose a seguir a EXP. O prazo de validade corresponde ao último dia do mês indicado.
Conserve os recipientes unidose na saqueta e na embalagem exterior, de forma a protegê-los da luz.
Use o recipiente unidose imediatamente após a abertura e rejeite após a utilização.
Após a abertura da saqueta, utilize os recipientes unidose no espaço de 1 mês.
Não deite fora quaisquer medicamentos na canalização. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de TIMOGEL
- A substância ativa é o timolol. 1 g de gel contém 1 mg de timolol sob a forma de maleato de timolol.
- Os outros componentes são o sorbitol, álcool polivinílico, carbómero 974P, acetato de sódio tri-hidratado, lisina mono-hidratada, água para preparações injetáveis.
Qual o aspeto de TIMOGEL e conteúdo da embalagem
TIMOGEL é um gel oftálmico opalescente, incolor ou ligeiramente amarelado, apresentado em recipientes unidose acondicionados em saquetas de 10 unidades; cada recipiente unidose contém 0,4 g de produto.
Uma embalagem contém 30 (3 x 10) ou 90 (9 x 10) recipientes unidose.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado:
LABORATOIRES THEA
12, RUE LOUIS BLERIOT
63017 CLERMONT- FERRAND CEDEX 2
FRANÇA
Fabricante:
LABORATOIRE UNITHER
ESPACE INDUSTRIEL NORD
151 RUE ANDRE DUROUCHEZ – BP 28028
80084 AMIENS CEDEX 2
FRANÇA
ou
LABORATOIRE UNITHER
1 rue de l’Arquerie
50200 COUTANCES
FRANÇA
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) com os seguintes nomes comerciais:
Holanda, Itália, Portugal: TIMOGEL
Bélgica, Bulgária, Grécia, Luxemburgo, Roménia: GELTIM
França: GELTIM LP
Irlanda: TIMOFLUID
Reino Unido: TIOPEX
Este folheto foi revisto pela última vez em 10/2021

Glaucoma
MONOPROST

Folheto informativo: Informação para o utilizador
MONOPROST 50 microgramas/ml colírio, solução em recipiente unidose
Latanoprost
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico, ou farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, ou farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
1. O que é MONOPROST e para que é utilizado
2. O que precisa de saber antes de utilizar MONOPROST
3. Como utilizar MONOPROST
4. Efeitos secundários possíveis
5. Como conservar MONOPROST
6. Conteúdo da embalagem e outras informações
1. O que é MONOPROST e para que é utilizado
MONOPROST pertence a um grupo de medicamentos designados por prostaglandinas. Reduz a pressão no interior do seu olho através do aumento da drenagem natural do líquido do interior do olho para a corrente sanguínea.
MONOPROST é utilizado para tratar doenças conhecidas por glaucoma de ângulo aberto e hipertensão ocular. Ambas as situações estão associadas a um aumento da pressão no interior do seu olho afetando, eventualmente, a sua visão.
2. O que precisa de saber antes de utilizar MONOPROST
Não utilize MONOPROST
- se tem alergia (hipersensibilidade) ao latanoprost ou a qualquer outro componente deste medicamento (indicados na secção 6).
Advertências e precauções
Fale com o seu médico, farmacêutico ou enfermeiro antes de utilizar MONOPROST, se pensa que alguma das seguintes situações lhe é aplicável:
• Se vai ser ou foi submetido a uma cirurgia aos olhos (incluindo cirurgia da catarata).
• Se sofre de problemas nos olhos (tais como dor, irritação ou inflamação nos olhos, visão turva).
• Se sofre de asma grave ou se a sua asma não está bem controlada.
• Se usa lentes de contacto. Pode utilizar MONOPROST, mas tem de seguir as instruções descritas na secção 3 para os utilizadores de lentes de contacto.
• Se tem ou já teve uma infeção viral nos olhos causada pelo vírus herpes simplex (VHS)
Crianças
MONOPROST não foi estudado em crianças (com menos de 18 anos).
Outros medicamentos e MONOPROST
MONOPROST pode interagir com outros medicamentos. Informe o seu médico ou farmacêutico se estiver a utilizar, tiver utilizado recentemente, ou se vier a utilizar outros medicamentos.
Gravidez e amamentação
Não utilize MONOPROST se está grávida ou a amamentar.
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de utilizar este medicamento.
Condução de veículos e utilização de máquinas
Quando utiliza MONOPROST poderá ficar com a visão turva, por um pequeno período de tempo. Se tal lhe acontecer, não conduza nem utilize ferramentas ou máquinas até que a sua visão fique novamente nítida.
MONOPROST contém hidroxi-estearato de macrogolglicerol
MONOPROST contém hidroxi-estearato de macrogolglicerol (derivado do óleo de castor) que pode causar reações na pele.
3. Como utilizar MONOPROST
Dose habitual
-Utilize MONOPROST exatamente como indicado pelo seu médico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
-A dose recomendada para adultos (incluindo idosos) é de uma gota no(s) olho(s) afetado(s), uma vez por dia. A melhor altura para efetuar a administração é à noite.
- Não utilize MONOPROST mais do que uma vez por dia, pois a eficácia do tratamento pode ser reduzida se administrar o medicamento com maior frequência.
- Utilize MONOPROST exatamente como descrito neste folheto ou como indicado pelo seu médico e até que o seu médico lhe diga para parar. Fale com o seu médico, farmacêutico ou enfermeiro se tiver dúvidas.
Utilizadores de lentes de contacto
Se utiliza lentes de contacto deve retirá-las antes de utilizar MONOPROST. Após a utilização de MONOPROST deve esperar 15 minutos antes de voltar a colocar as lentes.
Instruções de utilização
As gotas são fornecidas em recipientes unidose. A solução contida num recipiente unidose individual de MONOPROST deve ser utilizada imediatamente após a abertura para administração no(s) olho(s) afetado(s). Uma vez que a esterilidade não pode ser assegurada após a abertura do recipiente unidose individual, deve ser aberto um novo recipiente unidose antes de cada utilização e este deve ser rejeitado imediatamente após a administração.
Siga estas instruções para instilar as gotas:
1. Lave as suas mãos e sente-se ou coloque-se numa posição confortável.
2. Abra a saqueta. Escreva a data da primeira abertura da saqueta.
3. Separe um dos recipientes unidose do conjunto.

4. Rode o topo do recipiente unidose, como mostra a figura. Não toque na ponta do recipiente após a sua abertura.

5. Utilize o seu dedo para puxar cuidadosamente para baixo a pálpebra inferior do olho afetado.
6. Coloque a extremidade do recipiente unidose próximo do seu olho, mas de modo a não lhe tocar.
7. Pressione cuidadosamente o recipiente unidose de forma a que apenas uma gota entre no seu olho, depois liberte a pálpebra inferior.

8. Com um dedo pressione o canto do olho, mais próximo do nariz. Pressione durante 1 minuto, enquanto mantém o olho fechado.

9. Repita esta operação no outro olho, se o seu médico lhe tiver dito para o fazer.Cada recipiente unidose contém uma quantidade suficiente de solução para os dois olhos.
10. Rejeite o recipiente unidose após a utilização. Não conserve o recipiente para uma utilização posterior. Dado que a esterilidade não pode ser mantida após a abertura do recipiente unidose, deverá ser aberto um novo recipiente antes de cada utilização.
Se utilizar MONOPROST com outros colírios
Aguarde pelo menos 5 minutos entre a administração de MONOPROST e a administração de outros colírios.
Se utilizar mais MONOPROST do que deveria
Se administrar demasiadas gotas no seu olho poderá sentir uma ligeira irritação no seu olho e os seus olhos poderão ficar vermelhos e lacrimejantes. Esta situação deve passar, mas caso esteja preocupado fale com o seu médico.
Se ingerir MONOPROST acidentalmente, contacte o seu médico o mais rapidamente possível.
Caso se tenha esquecido de utilizar MONOPROST
Aplique a dose habitual na devida altura. Não aplique uma dose a dobrar para compensar a dose que se esqueceu de aplicar. Caso tenha dúvidas contacte o seu médico ou farmacêutico.
Se parar de utilizar MONOPROST
Deve falar com o seu médico se pretende parar de utilizar MONOPROST.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico, farmacêutico ou enfermeiro.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Os seguintes efeitos secundários são conhecidos por ocorrer com a utilização de MONOPROST.
Muito frequentes: podem afetar mais de 1 em 10 pessoas
• Alteração gradual na cor dos olhos pelo aumento do pigmento castanho na parte colorida do olho denominada íris.
- Se tem olhos com mais do que uma cor (azuis-castanhos, cinzentos-castanhos, amarelos-castanhos, verdes-castanhos) tem maior probabilidade de observar esta alteração do que se tiver olhos de cor única (olhos azuis, cinzentos, verdes ou castanhos).
- Qualquer alteração na cor dos seus olhos pode levar anos a ocorrer, embora seja geralmente observada nos primeiros 8 meses de tratamento.
- A alteração da cor pode ser permanente e pode ser mais evidente caso utilize MONOPROST em apenas um olho.
- Parece não existirem problemas associados com a alteração da cor do(s) olho(s).
- A alteração de cor não se mantém após a interrupção do tratamento com MONOPROST.
• Vermelhidão no olho.
• Irritação no olho (sensação de queimadura, de areia nos olhos, comichão, picadas ou sensação de corpo estranho no olho).
• Alteração gradual das pestanas no olho tratado e dos finos pelos à volta do olho tratado, observada essencialmente em pessoas de origem Japonesa. Estas alterações incluem um aumento da cor (escurecimento), comprimento, espessura e número das suas pestanas.
Frequentes: podem afetar até 1 em 10 pessoas
• Irritação ou alteração na superfície do olho, inflamação das pálpebras (blefarite), dor nos olhos, sensibilidade à luz (fotofobia), conjuntivite.
Pouco frequentes: podem afetar até 1 em 100 pessoas
• Pálpebras inchadas, secura do olho, inflamação ou irritação da superfície do olho (queratite), visão turva, inflamação da parte colorida do olho (uveíte), inchaço da retina (edema macular).
• Erupção na pele.
• Dor no peito (angina), perceção do ritmo cardíaco (palpitações).
• Asma, falta de ar (dispneia).
• Dor no peito.
• Dores de cabeça, tonturas.
• Dores nos músculos, dores nas articulações.
• Náuseas, vómitos.
Raros: podem afetar até 1 em 1.000 pessoas
• Inflamação da íris (irite), sintomas de inchaço ou escoriação/lesão da superfície do olho, inchaço à volta do olho (edema periorbital), pestanas na “direção errada” ou uma fila “extra” de pestanas, zona cheia de líquido na parte colorida do olho (quistos na íris).
• Reações na pele das pálpebras, escurecimento da pele das pálpebras.
• Agravamento da asma.
• Comichão grave na pele.
• Desenvolvimento de uma infeção viral no olho causada pelo vírus herpes simplex (VHS).
Muito raros: podem afetar até 1 em 10.000 pessoas
• Agravamento da angina em doentes que tenham também doença cardíaca, olhos com aspeto encovado (aprofundamento do sulco da pálpebra)
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente através do sistema nacional de notificação mencionado abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Fax: +351 21 798 73 97
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar MONOPROST
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior, saqueta e recipiente unidose. O prazo de validade corresponde ao último dia do mês indicado.
Conservar abaixo de 25ºC.
Após a primeira abertura da saqueta: utilizar os recipientes unidose no prazo de 10 dias.
Após a primeira abertura do recipiente unidose: utilizar imediatamente e rejeitar o recipiente unidose após a utilização.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de MONOPROST
- A substância ativa é o latanoprost.
1 ml de colírio, solução contém 50 microgramas de latanoprost.
- Os outros componentes são: hidroxi-estearato de macrogolglicerol 40, sorbitol, carbómero 974P, macrogol 4000, edetato dissódico, hidróxido de sódio (para ajuste do pH) e água para preparações injetáveis.
Qual o aspeto de MONOPROST e conteúdo da embalagem
Este medicamento apresenta-se sob a forma de colírio, solução em recipiente unidose. A solução é ligeiramente amarelada e opalescente, sem conservantes, contida em recipientes unidose acondicionados numa saqueta com 5 ou 10 unidades; cada recipiente unidose contém 0,2 ml de produto.
Cada embalagem contém 5 (1x5), 10 (2x5), 10 (1x10), 30 (6x5), 30 (3x10), 90 (18x5) ou 90 (9x10) recipientes unidose.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires Théa
12, Rue Louis Blériot
Zone Industrielle du Brézet
63017 Clermont-Ferrrand
França
Fabricante
EXCELVISION
27, rue de la Lombardière
07100 Annonay
França
Ou
Laboratoires Théa
12 Rue Louis Blériot
Zone Industrielle du Brézet
63017 Clermont-Ferrrand
França
Ou
LABORATOIRE UNITHER
ZI de la Guérie
50211 Coutances Cedex
França
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Alemanha, Bélgica, Bulgária, Chipre, Dinamarca, Espanha, Estónia, Finlândia, França, Grécia, Holanda, Itália, Islândia, Letónia, Luxemburgo, Noruega, Polónia, Portugal, Suécia..............Monoprost
Irlanda......................................................................................................................Monopost Unidose
Áustria, Eslováquia, Eslovénia, Lituânia, República Checa, Reino Unido, Roménia................Monopost
Estónia…………………………………………………………………………………… Monopro
Este folheto foi revisto pela última vez em 02/2024

Glaucoma
Duokopt

Folheto informativo: Informação para o utilizador
Duokopt 5 mg/ml + 20 mg/ml colírio, solução
Timolol + Dorzolamida
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico, farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
1. O que é Duokopt e para que é utilizado
2. O que precisa de saber antes de utilizar Duokopt
3. Como utilizar Duokopt
4. Efeitos secundários possíveis
5. Como conservar Duokopt
6. Conteúdo da embalagem e outras informações
1. O que é Duokopt e para que é utilizado
Duokopt é um colírio, solução sem conservantes.
Duokopt contém duas substâncias ativas: timolol e dorzolamida.
- A dorzolamida pertence a um grupo de medicamentos denominados “inibidores da anidrase carbónica”.
- O timolol pertence a um grupo de medicamentos denominados “bloqueadores beta”.
Estes medicamentos reduzem a pressão no olho de diferentes formas.
Duokopt é prescrito para reduzir a pressão elevada no olho no tratamento do glaucoma quando não é adequado utilizar, isoladamente, um colírio com um bloqueador beta.
2. O que precisa de saber antes de utilizar Duokopt
Não utilize Duokopt:
- se tem alergia (hipersensibilidade) ao cloridrato de dorzolamida, ao maleato de timolol ou a qualquer outro componente deste medicamento (indicados na secção 6).
- se tem ou já teve problemas respiratórios tais como asma, bronquite obstrutiva crónica grave (doença pulmonar grave que pode causar pieira, dificuldade em respirar e/ou tosse prolongada).
- se tem um ritmo cardíaco lento, insuficiência cardíaca ou alterações do ritmo cardíaco (batimento cardíaco irregular).
- se tem problemas ou doenças graves nos rins ou antecedentes de pedras nos rins.
- se tem excessiva acidez do sangue causada por um aumento de cloretos no sangue
(acidose hiperclorémica).
Se não tem a certeza se pode utilizar Duokopt contacte o seu médico ou farmacêutico.
Advertências e precauções
Fale com o seu médico, farmacêutico ou enfermeiro antes de utilizar Duokopt acerca de quaisquer problemas de saúde ou problemas nos seus olhos que tenha agora ou que já teve no passado:
- doença cardíaca coronária (os sintomas podem incluir dor ou aperto no peito, falta de ar ou sufoco), insuficiência cardíaca, pressão arterial baixa.
- alterações do ritmo cardíaco tais como ritmo cardíaco lento.
- problemas respiratórios, asma ou doença pulmonar obstrutiva crónica.
- problemas na circulação sanguínea (como doença de Raynaud ou síndrome de Raynaud).
- diabetes, uma vez que o timolol pode mascarar os sinais e sintomas de níveis baixos de açúcar no sangue.
- hiperatividade da glândula tiroide, uma vez que o timolol pode mascarar sinais e sintomas.
- quaisquer alergias ou reações anafiláticas.
- fraqueza muscular, especialmente se lhe tiver sido diagnosticada miastenia gravis.
Antes de uma cirurgia, informe o seu médico que está a tomar Duokopt pois o timolol pode alterar o efeito de alguns medicamentos utilizados durante a anestesia.
Durante o tratamento com Duokopt contacte o seu médico imediatamente se:
- desenvolver qualquer irritação nos olhos ou outros problemas nos olhos tais como vermelhidão nos olhos ou inchaço nas pálpebras.
- suspeitar que Duokopt lhe está a causar uma reação alérgica ou hipersensibilidade (por exemplo, erupções na pele, reação grave na pele ou vermelhidão e comichão no olho), pare de utilizar Duokopt.
- desenvolver uma infeção no olho, sofrer um ferimento no olho, for submetido a uma cirurgia aos olhos ou desenvolver uma reação com novos sintomas ou agravamento dos sintomas.
A aplicação de Duokopt no olho pode afetar todo o organismo.
Caso utilize lentes de contacto
Duokopt não foi estudado em doentes que utilizam lentes de contacto.
Caso utilize lentes de contacto, deve consultar o seu médico antes de utilizar Duokopt.
Crianças
A experiência de utilização da associação timolol + dorzolamida com conservantes em lactentes e crianças é limitada. Duokopt não é recomendado em crianças com idades entre os 0 e os 18 anos.
Idosos
Nos estudos realizados com a associação timolol + dorzolamida com conservantes, os efeitos deste medicamento foram semelhantes tantos nos idosos como nos doentes mais jovens.
Doentes com insuficiência da função do fígado
Informe o seu médico sobre quaisquer problemas no fígado que tenha ou que tenha tido no passado.
Doentes com insuficiência da função dos rins
Informe o seu médico sobre quaisquer problemas nos rins que tenha ou que tenha tido no passado.
Desporto
A utilização de Duokopt pode provocar resultados positivos nos controlos anti-doping.
Outros medicamentos e Duokopt
O Duokopt pode afetar ou ser afetado por outros medicamentos que esteja a utilizar, incluindo outros colírios para o tratamento do glaucoma.
Informe o seu médico ou farmacêutico se estiver a utilizar, tiver utilizado recentemente, ou se vir a utilizar outros medicamentos.
Isto é especialmente importante se está a:
- tomar medicamentos para baixar a pressão arterial ou para tratar doenças cardíacas
(tais como os bloqueadores dos canais de cálcio, bloqueadores beta ou digoxina).
- tomar medicamentos para tratar batimentos cardíacos alterados ou irregulares, tais como os bloqueadores dos canais de cálcio, bloqueadores beta ou digoxina.
- utilizar outro colírio para os olhos que contenha um bloqueador beta
- tomar outro inibidor da anidrase carbónica, tal como a acetazolamida.
- tomar inibidores da monoamino oxidase (IMAO), os quais são utilizados para tratar a depressão.
- tomar um medicamento parassimpaticomimético que lhe possa ter sido prescrito para o ajudar a urinar. Os parassimpaticomiméticos são também um tipo particular de medicamento que por vezes são utilizados para ajudar a restabelecer os movimentos normais do intestino.
- tomar narcóticos tal como a morfina, que é utilizada para tratar dores moderadas a graves.
- tomar medicamentos para tratar a diabetes.
- tomar antidepressivos, conhecidos como fluoxetina e paroxetina.
- tomar um medicamento sulfa.
- tomar quinidina (utilizada para tratar doenças cardíacas e alguns tipos de malária).
Gravidez e amamentação
Não utilize Duokopt se está grávida a não ser que o seu médico considere necessário.
Consulte o seu médico ou farmacêutico para obter aconselhamento antes de tomar qualquer medicamento.
Não utilize Duokopt se está a amamentar. O timolol pode passar para o leite materno. Consulte o seu médico para obter aconselhamento sobre a toma de qualquer medicamento durante a amamentação.
Condução de veículos e utilização de máquinas
Não foram realizados estudos sobre os efeitos na capacidade de conduzir ou utilizar máquinas. Existem efeitos secundários associados ao Duokopt, tais como visão turva, que podem afetar a sua capacidade para conduzir e/ou utilizar máquinas. Não conduza nem utilize máquinas até se sentir bem ou sentir que a visão voltou a estar límpida.
3. Como utilizar Duokopt
Utilize este medicamento exatamente como indicado pelo seu médico. Fale com o seu médico ou farmacêutico se tiver dúvidas. O seu médico irá estabelecer a dose e a duração do tratamento apropriadas.
Posologia
- A dose habitual é de uma gota no(s) olho(s) afetado(s) de manhã e à noite.
- Se está a utilizar Duokopt com outro colírio, as gotas devem ser administradas com, pelo menos, 10 minutos de intervalo.
- Não altere a dose do medicamento sem consultar o seu médico.
- Não deixe que a ponta do frasco toque no olho ou na zona à volta do olho. Este pode ficar contaminado com bactérias que podem causar infeções nos olhos originando graves lesões nos olhos, ou mesmo perda de visão. Para evitar qualquer possível contaminação do frasco, mantenha a ponta do frasco afastada de qualquer superfície.
Instruções de utilização
Quando utilizar o medicamento é importante que siga as seguintes instruções:
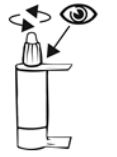 |
Antes de utilizar o frasco pela primeira vez, certifique-se de que a tampa de segurança não está danificada. De seguida desenrosque com firmeza a tampa de segurança para abrir o frasco. |
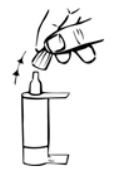 |
1. Antes de cada utilização, lave cuidadosamente as suas mãos e retire a tampa da ponta do frasco. Evite tocar na ponta do frasco com os dedos. |
| Para ativar o doseador, com o frasco virado para baixo, pressione várias vezes o frasco até que apareça a primeira gota. Este passo é apenas necessário na primeira utilização, não sendo necessário repeti-lo nas administrações seguintes. | |
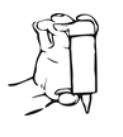 |
2. Coloque o polegar na aba da parte superior do frasco e o dedo indicador na base do frasco. Em seguida coloque também o dedo médio na segunda aba que está na base do frasco. Segure no frasco com a ponta virada para baixo. |
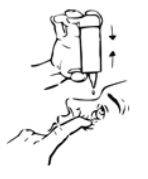 |
3. Para utilizar, incline a sua cabeça ligeiramente para trás e coloque o doseador na vertical sobre o seu olho. Com o dedo indicador da outra mão, puxe a pálpebra inferior ligeiramente para baixo. O espaço criado designa-se por saco conjuntival inferior. Evite tocar na ponta do frasco com os seus dedos ou com os seus olhos. Para aplicar uma gota no saco conjuntival inferior do(s) olho(s) afetado(s), pressione momentaneamente mas com firmeza o frasco. Devido ao doseador automático, é libertada uma gota exatamente após cada atuação. |
| Se não cair qualquer gota, agite suavemente o frasco para remover a gota da ponta do frasco. Nesta situação repita o passo 3. | |
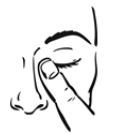 |
Feche a pálpebra e pressione com o seu dedo o canto interior do olho junto ao nariz durante 2 minutos. Tal ajuda a prevenir que o colírio se espalhe para o resto do seu corpo. |
| Feche o frasco com a tampa imediatamente após a utilização. | |
Se utilizar mais Duokopt do que deveria
Se aplicar demasiadas gotas nos seus olhos ou se engolir o líquido que está no frasco, poderá, entre outros efeitos, sentir-se atordoado, com dificuldade em respirar, ou sentir que o seu ritmo cardíaco diminuiu. Contacte imediatamente o seu médico.
Caso se tenha esquecido de utilizar Duokopt
É importante que utilize o Duokopt conforme prescrito pelo seu médico.
Caso se tenha esquecido de uma dose, aplique-a assim que possível. No entanto, se estiver quase na hora de aplicar a dose seguinte, não aplique a dose em atraso e volte a aplicar as gotas dentro do horário previsto.
Não aplique uma dose a dobrar para compensar uma dose que se esqueceu de aplicar.
Se parar de utilizar Duokopt
Se pretende parar de utilizar este medicamento, fale primeiro com o seu médico.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico, farmacêutico ou enfermeiro.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
De um modo geral poderá continuar a administrar as suas gotas, exceto se os efeitos forem graves. Se estiver preocupado, contacte o seu médico ou farmacêutico. Não pare de utilizar Duokopt sem consultar o seu médico.
Reações alérgicas
Podem ocorrer reações alérgicas generalizadas, incluindo inchaço sob a pele, em áreas tais como a face e os membros, que podem obstruir as vias aéreas causando dificuldades em engolir ou em respirar, urticária ou erupção na pele com comichão, erupção na pele localizada ou generalizada, comichão, ou reação alérgica grave e súbita que pode colocar a vida em risco. Se tiver algum destes efeitos, pare de utilizar Duokopt e contacte imediatamente o seu médico.
Muito frequentes: podem afetar mais de 1 em 10 pessoas
Sensação de queimadura e picadas nos olhos, alterações do paladar.
Frequentes: podem afetar até 1 em 10 pessoas
Vermelhidão no(s) olho(s) e zonas circundantes, lacrimejo ou comichão no(s) olho(s), erosão da córnea (lesão na primeira camada do globo ocular) , inchaço e/ou irritação no(s) olho(s) e zonas circundantes, sensação de objeto estranho no olho, sensibilidade da córnea diminuída (não ter consciência de ter um objeto estranho no olho e não sentir dor), dor nos olhos, olhos secos, visão turva, dor de cabeça, sinusite (sensação de tensão ou obstrução no nariz), náuseas, fraqueza/cansaço e fadiga.
Pouco frequentes: podem afetar até 1 em 100 pessoas
Tonturas, depressão, inflamação da íris, perturbações visuais incluindo alterações da refração (em alguns casos devido à interrupção da terapêutica miótica), diminuição do ritmo cardíaco, desmaios, dificuldade em respirar (dispneia), indigestão e pedras nos rins.
Raros: podem afetar até 1 em 1.000 pessoas
Lúpus eritematoso sistémico (uma doença imunitária que pode causar inflamação dos órgãos internos), formigueiro ou dormência das mãos e pés, insónias, pesadelos, perda de memória, um aumento dos sinais e sintomas de miastenia gravis (doença muscular), diminuição do apetite sexual, acidente vascular cerebral, miopia temporária que pode resolver-se com a interrupção do tratamento, descolamento da membrana abaixo da retina onde se localizam os vasos sanguíneos após cirurgia de filtração, que pode provocar alterações da visão, pálpebras descaídas (o que faz com que olho fique semifechado), visão dupla, crostas nas pálpebras, inchaço da córnea (com sintomas de alterações visuais), pressão diminuída no olho, zumbidos nos ouvidos, pressão arterial baixa, alterações no ritmo ou na velocidade do batimento cardíaco, insuficiência cardíaca congestiva (doença cardíaca com sintomas de falta de ar e inchaço dos pés e pernas provocado por acumulação de líquidos), edema (acumulação de líquidos), isquemia cerebral (aporte de sangue reduzido para o cérebro), dor no peito, batimentos cardíacos fortes que podem ser rápidos ou irregulares (palpitações), ataque cardíaco, fenómeno de Raynaud, inchaço ou arrefecimento das mãos e pés e diminuição da circulação nos braços e pernas, cãibras nas pernas e/ou dor nas pernas ao andar (claudicação), falta de ar, insuficiência respiratória, rinite, hemorragia nasal, constrição das vias aéreas nos pulmões, tosse, irritação na garganta, boca seca, diarreia, dermatite de contacto, perda de cabelo, erupção na pele com aparência branca-prateada (psoríase), doença de Peyronie (que pode causar uma curvatura no pénis), reações de tipo alérgico como erupção cutânea, urticária, comichão, em casos raros possível inchaço dos lábios, olhos e boca, pieira ou reações graves na pele (síndrome de Stevens Johnson, necrólise epidérmica tóxica).
Tal como com outros medicamentos de aplicação nos olhos, o timolol é absorvido para o sangue. Tal poderá provocar efeitos secundários semelhantes aos observados com bloqueadores beta administrados por via oral. A incidência de efeitos secundários após administração oftalmológica tópica é inferior à observada quando, por exemplo, os medicamentos são administrados por via oral ou injetável.
Os efeitos secundários seguintes foram notificados com medicamentos do mesmo grupo, quando utilizados para o tratamento de doenças dos olhos.
Desconhecida: a frequência não pode ser estimada a partir dos dados disponíveis
Níveis de glucose no sangue diminuídos, insuficiência cardíaca, um tipo de alteração do ritmo cardíaco, dor abdominal, vómitos, dor muscular não provocada por exercício, disfunção sexual, falta de ar, sensação de corpo estranho no olho (sensação de que está alguma coisa no seu olho), alucinação.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Fax: +351 21 798 73 97
Sítio da internet: http://extranet.infarmed.pt/page.seram.frontoffice.seramhomepage
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar Duokopt
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior e no frasco após EXP. O prazo de validade corresponde ao último dia do mês indicado.
Este medicamento não necessita de quaisquer precauções especiais de conservação.
O Duokopt pode ser utilizado até 2 meses após a primeira abertura do frasco. Escreva a data da primeira abertura na embalagem.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Duokopt
- As substâncias ativas são: timolol e dorzolamida. Cada ml contém 5 mg de timolol (sob a forma de 6,83 mg de maleato de timolol) e 20 mg de dorzolamida (sob a forma de 22,25 mg de cloridrato de dorzolamida).
- Os outros componentes são: hidroxietilcelulose, manitol, citrato de sódio, hidróxido de sódio para ajuste do pH e água para preparações injetáveis.
Qual o aspeto de Duokopt e conteúdo da embalagem
Duokopt é uma solução límpida incolor ou ligeiramente amarelada.
O Duokopt encontra-se disponível nas seguintes apresentações:
- frasco multidose de 5 ml (com pelo menos 125 gotas sem conservantes, para 1 mês de tratamento), incluindo um doseador com um dispositivo que permite um manuseamento ergonómico.
- frasco multidose de 10 ml (com pelo menos 250 gotas sem conservantes, para 2 meses de tratamento), incluindo um doseador com um dispositivo que permite um manuseamento ergonómico.
- 1 embalagem com três frascos multidose de 5 ml (3 meses de tratamento) incluindo cada frasco um doseador com um dispositivo que permite um manuseamento ergonómico.
- 3 embalagens cada uma com um frasco multidose de 5 ml acondicionadas com película (3 meses de tratamento) incluindo cada frasco um doseador com um dispositivo que permite um manuseamento ergonómico.
- 1 embalagem com dois frascos multidose de 10 ml (4 meses de tratamento) incluindo cada frasco um doseador com um dispositivo que permite um manuseamento ergonómico.
- 2 embalagens cada uma com um frasco multidose de 10 ml acondicionadas com película (4 meses de tratamento) incluindo cada frasco um doseador com um dispositivo que permite um manuseamento ergonómico.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires Théa
12 rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
Fabricante
Delpharm Tours
Rue Paul Langevin
37170 Chambray les Tours
França
ou
LaboratoiresThéa
12 rue Louis Blériot
63017 Clermont-FerrandCedex 2
França
ou
FARMILA-THEA Farmaceutici S.p.A.
Via Enrico Fermi, 50
20019 Settimo Milanese
Itália
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Alemanha, Áustria, Bulgária, Chipre, Dinamarca, Eslovénia, Espanha, Grécia, Finlândia, Islândia, Itália, Noruega, Polónia, Portugal, Roménia, Suécia …………………………..Duokopt
Bélgica, Eslováquia, França, Holanda, Luxemburgo, República Checa …………….Dualkopt
Este folheto foi revisto pela última vez em 09/2023

Glaucoma
MONOPROST DUO

Folheto informativo: Informação para o doente
Monoprost Duo 50 microgramas/ml + 5 mg/ml colírio, solução em recipiente unidose
latanoprost + timolol
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico, ou farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico, ou farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
1. O que é Monoprost Duo e para que é utilizado
2. O que precisa de saber antes de utilizar Monoprost Duo
3. Como utilizar Monoprost Duo
4. Efeitos secundários possíveis
5. Como conservar Monoprost Duo
6. Conteúdo da embalagem e outras informações
1. O que é Monoprost Duo e para que é utilizado
Monoprost Duo contém dois medicamentos: latanoprost e timolol. Latanoprost pertence a um grupo de medicamentos designado por análogos das prostaglandinas. Timolol pertence a um grupo de medicamentos designado por bloqueadores beta. O latanoprost atua aumentando a drenagem natural de líquido do interior do olho para a corrente sanguínea. O timolol atua diminuindo a formação de líquido no olho.
Monoprost Duo é utilizado para reduzir a pressão no seu olho, em situações como o glaucoma de ângulo aberto ou hipertensão ocular. Ambas as situações estão associadas a um aumento de pressão no interior do seu olho afetando, eventualmente, a sua visão. O seu médico irá geralmente prescrever-lhe Monoprost Duo quando outros medicamentos não forem suficientemente eficazes.
2. O que precisa de saber antes de utilizar Monoprost Duo
Monoprost Duo pode ser utilizado em homens e mulheres adultos (incluindo idosos), mas não se recomenda a sua utilização se tiver menos de 18 anos de idade.
Não utilize Monoprost Duo:
- se tem alergia (hipersensibilidade) ao latanoprost, timolol, bloqueadores beta ou a qualquer outro componente deste medicamento (indicados na secção 6),
- se tem ou teve no passado problemas respiratórios tais como asma, bronquite obstrutiva crónica grave (doença grave nos pulmões que pode causar respiração ofegante, dificuldade em respirar e/ou tosse permanente),
- se tem problemas graves no coração ou alterações do ritmo cardíaco.
Advertências e precauções
Fale com o seu médico, farmacêutico ou enfermeiro antes de utilizar Monoprost Duo, se tem ou teve no passado:
- doença cardíaca coronária (os sintomas podem incluir dor ou aperto no peito, falta de ar ou asfixia), insuficiência cardíaca, pressão arterial baixa,
- perturbações no ritmo cardíaco, tais como diminuição dos batimentos cardíacos,
- problemas respiratórios, asma ou doença pulmonar obstrutiva crónica,
- doença de má circulação do sangue (como a doença de Raynaud ou síndrome de Raynaud),
- diabetes, uma vez que o timolol pode mascarar sinais e sintomas de níveis baixos de açúcar no sangue,
- hiperatividade da glândula tiroide, uma vez que o timolol pode mascarar os seus sinais e sintomas,
- se vai ser submetido a qualquer tipo de cirurgia aos olhos (incluindo cirurgia da catarata) ou se no passado foi submetido a qualquer tipo de cirurgia aos olhos,
- se sofre de problemas nos olhos (tais como dor nos olhos, irritação nos olhos, inflamação nos olhos ou visão turva),
- se sabe que sofre de olho seco,
- se usa lentes de contacto. Pode utilizar Monoprost Duo, mas tem de seguir as instruções descritas na secção 3 para os utilizadores de lentes de contacto,
- se sabe que sofre de angina (em particular um tipo de angina conhecido como angina de Prinzmetal),
- se sabe que sofre de reações alérgicas graves que usualmente necessitam de tratamento hospitalar,
- se tem ou já teve uma infeção viral nos olhos causada pelo vírus do herpes simplex (VHS).
Informe o seu médico que está a utilizar Monoprost Duo antes de ser operado, uma vez que o timolol pode alterar os efeitos de alguns medicamentos utilizados durante a anestesia.
Outros medicamentos e Monoprost Duo
Informe o seu médico ou farmacêutico se estiver a tomar, tiver tomado recentemente, ou se vier a tomar outros medicamentos, incluindo colírios e medicamentos obtidos sem receita médica.
Monoprost Duo pode afetar ou ser afetado por outros medicamentos que esteja a tomar, incluindo outros colírios para o tratamento do glaucoma. Informe o seu médico se está a tomar ou tenciona tomar medicamentos para baixar a pressão arterial, medicamentos para o coração ou medicamentos para a diabetes.
É importante que informe o seu médico ou farmacêutico se está a tomar algum dos seguintes tipos de medicamentos:
- Prostaglandinas, análogos das prostaglandinas ou derivados das prostaglandinas,
- Bloqueadores beta,
- Epinefrina,
- Medicamentos para tratar a pressão arterial alta tais como bloqueadores dos canais de cálcio, guanetidina, antiarrítmicos, glicosídeos digitálicos ou parassimpaticomiméticos,
- Quinidina (utilizada para o tratamento de doenças cardíacas e alguns tipos de malária),
- Antidepressivos conhecidos como fluoxetina e paroxetina.
Monoprost Duo com alimentos e bebidas:
Refeições normais, alimentos ou bebidas não têm qualquer efeito em quando ou como deve utilizar Monoprost Duo.
Gravidez, amamentação e fertilidade
Não utilize Monoprost Duo se estiver grávida ou a amamentar.
Monoprost Duo pode passar para o leite materno.
O latanoprost e o timolol não demonstraram ter qualquer efeito na fertilidade masculina ou feminina em estudos em animais.
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico antes de utilizar este medicamento.
Condução de veículos e utilização de máquinas
Quando utiliza Monoprost Duo poderá ficar com a visão turva, por um breve período de tempo. Se isto lhe acontecer, não conduza nem utilize ferramentas ou máquinas até que a sua visão fique novamente nítida.
Monoprost Duo contém hidroxi-estearato de macrogolglicerol (derivado do óleo de castor) que pode causar reações na pele.
3. Como utilizar Monoprost Duo
Utilize este medicamento exatamente como indicado pelo seu médico ou farmacêutico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
A dose recomendada para adultos (incluindo os idosos) é de uma gota no(s) olho(s) afetado(s), uma vez por dia.
Não utilize Monoprost Duo mais do que uma vez por dia, pois a eficácia do tratamento pode ser reduzida se administrar o medicamento com maior frequência.
Utilize Monoprost Duo como indicado pelo seu médico e até que o seu médico lhe diga para parar.
Enquanto estiver a utilizar Monoprost Duo, o seu médico pode querer que faça exames adicionais ao coração e circulação.
Utilizadores de lentes de contacto
Se usa lentes de contacto deve retirá-las antes de utilizar Monoprost Duo. Após a aplicação de Monoprost Duo deve esperar 15 minutos antes de voltar a colocar as suas lentes.
Instruções de utilização
Este medicamento destina-se a ser administrado no olho.
Siga por favor estas instruções para utilizar as gotas:
1. Lave as suas mãos e sente-se ou coloque-se numa posição confortável.
2. Abra a saqueta que contém 5 recipientes unidose. Aponte a data da primeira abertura na saqueta.
3. Separe um dos recipientes unidose do conjunto.
1. Lave as suas mãos e sente-se ou coloque-se numa posição confortável.
2. Abra a saqueta que contém 5 recipientes unidose. Aponte a data da primeira abertura na saqueta.
3. Separe um dos recipientes unidose do conjunto.

4.Para abrir, rode o topo do recipiente unidose, como mostra a figura. Não toque na ponta do recipiente após a sua abertura.

5.Utilize o seu dedo para puxar cuidadosamente para baixo a pálpebra inferior do seu olho afetado.
6.Coloque a extremidade do recipiente unidose próximo do seu olho, mas de modo a não lhe tocar.
7.Pressione cuidadosamente o recipiente unidose de forma a que apenas uma gota entre no seu olho, depois liberte a pálpebra inferior.

8. Com um dedo pressione o canto do olho mais próximo do nariz. Pressione durante 2 minutos, mantendo o olho fechado.

9.Repita no outro olho, se o seu médico lhe tiver dito para o fazer. Cada recipiente unidose contém uma quantidade suficiente de solução para os dois olhos.
10.Rejeite o recipiente unidose após a utilização. Não conserve o recipiente para o utilizar novamente. Dado que a esterilidade não pode ser mantida após a abertura do recipiente unidose, deverá ser aberto um novo recipiente antes de cada utilização.
11.Volte a colocar dentro da saqueta os recipientes unidose não abertos. Coloque a saqueta aberta dentro da embalagem original. Os restantes recipientes unidose devem ser utilizados até 1 mês após a abertura da saqueta.
Se utilizar Monoprost Duo com outros colírios
Aguarde pelo menos 5 minutos entre a administração de Monoprost Duo e a administração de outros colírios.
Se utilizar mais Monoprost Duo do que deveria
Se administrar demasiadas gotas no seu olho poderá sentir uma ligeira irritação no seu olho e os seus olhos poderão ficar vermelhos e lacrimejantes. Esta situação deve passar, mas caso esteja preocupado fale com o seu médico.
Se ingerir Monoprost Duo
Se ingerir Monoprost Duo acidentalmente, deverá contactar o seu médico assim que possível. Se ingeriu uma grande quantidade de Monoprost Duo pode sentir-se enjoado, ter dores de estômago, sentir-se cansado, corado, com tonturas e começar a transpirar.
Caso se tenha esquecido de utilizar Monoprost Duo
Aplique a dose habitual na devida altura. Não utilize uma dose a dobrar para compensar a dose que se esqueceu de utilizar. Caso tenha dúvidas contacte o seu médico ou farmacêutico.
4. Efeitos secundários possíveis
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Pode continuar a utilizar as gotas, a menos que os efeitos sejam graves. Se estiver preocupado, fale com o seu médico ou farmacêutico. Não deixe de utilizar Monoprost Duo sem falar com o seu médico.
Os efeitos secundários conhecidos da utilização de Monoprost Duo encontram-se listados abaixo. O efeito secundário mais importante é a possibilidade de uma alteração gradual e permanente na cor do olho. Também é possível que Monoprost Duo possa causar alterações graves no funcionamento do seu coração. Se notar alterações na frequência cardíaca ou no funcionamento do coração, deve falar com o seu médico e informá-lo/informá-la que está a utilizar Monoprost Duo.
Os efeitos secundários conhecidos da utilização de Monoprost Duo são os seguintes:
Muito frequentes (podem afetar mais de 1 em 10 pessoas):
- Alteração gradual na cor dos olhos pelo aumento do pigmento castanho na parte colorida do olho denominada íris. Se tem olhos com mais do que uma cor (azuis-castanhos, cinzentos castanhos, amarelos-castanhos ou verdes-castanhos) tem maior probabilidade de observar esta alteração do que se tiver olhos de cor única (olhos azuis, cinzentos, verdes ou castanhos). Qualquer alteração na cor dos seus olhos pode levar anos a ocorrer. A alteração da cor pode ser permanente e pode ser mais evidente caso utilize Monoprost Duo apenas num dos olhos. Parece não existirem problemas associados com a alteração da cor dos olhos. A alteração da cor não continua após a interrupção do tratamento com Monoprost Duo.
Frequentes (podem afetar até 1 em 10 pessoas):
-
Irritação no olho (sensação de ardor, areia nos olhos, comichão, picadas ou sensação de corpo estranho no olho) e dor no olho.
Pouco frequentes (podem afetar até 1 em 100 pessoas):
-
Dor de cabeça,
-
Vermelhidão no olho, infeção no olho (conjuntivite), visão turva, olhos lacrimejantes, inflamação das pálpebras, irritação ou perturbação na superfície do olho,
- Erupção na pele ou comichão (prurido),
- Náuseas, vómitos.
Outros efeitos secundários
Tal como outros medicamentos aplicados nos olhos, Monoprost Duo (latanoprost e timolol) é absorvido para o sangue. A incidência de efeitos secundários após a utilização de colírios é menor do que quando esses medicamentos são administrados, por exemplo, por via oral ou injetável.
Embora não observados com Monoprost Duo, os seguintes efeitos secundários adicionais têm sido observados com as substâncias ativas de Monoprost Duo (latanoprost e timolol) e por isso, podem ocorrer quando utilizar Monoprost Duo.
Os efeitos secundários listados incluem reações observadas com a classe dos bloqueadores beta (p. ex. timolol) quando utilizados para tratamento de doenças do olho:
- Desenvolvimento de uma infeção viral no olho causada pelo vírus herpes simplex (VHS).
- Reações alérgicas generalizadas incluindo inchaço debaixo da pele que pode ocorrer em zonas como a face e membros e pode obstruir as vias aéreas, o que pode causar dificuldade em engolir ou respirar, urticária ou erupção na pele com comichão, erupção na pele localizada e generalizada, comichão, reação alérgica aguda grave com risco de vida.
- Níveis baixos de glicose no sangue.
- Tonturas.
- Dificuldade em dormir (insónia), depressão, pesadelos, perda de memória, alucinação.
- Desmaio, acidente vascular cerebral, diminuição do fornecimento de sangue ao cérebro, aumento dos sinais e sintomas de miastenia gravis (doença dos músculos), sensação esquisita de picadas de agulhas e alfinetes e dor de cabeça.
- Inchaço da parte posterior do olho (edema macular), quisto cheio de líquido na parte colorida do olho (quisto na íris), sensibilidade à luz (fotofobia), aspeto de olhos encovados (aprofundamento do sulco do olho).
- Sinais e sintomas de irritação ocular (p. ex. ardor, picadas, comichão, lacrimejo, vermelhidão), inflamação da pálpebra, inflamação na córnea, visão turva e descolamento da camada abaixo da retina que contém vasos sanguíneos após cirurgia de filtração, o que pode causar alterações da visão, diminuição da sensibilidade da córnea, olhos secos, erosão da córnea (danos na camada da frente do globo ocular), pálpebra superior descaída (ficando o olho meio fechado), visão dupla.
- Escurecimento da pele em torno dos olhos, alterações das pestanas e dos finos pelos à volta do olho (aumento do número, comprimento, espessura e escurecimento), alterações na direção do crescimento das pestanas, inchaço à volta do olho, inchaço da parte colorida do olho (irite/uveíte), pequenas cicatrizes na superfície do olho.
- Assobios/zumbidos no ouvido (tinidos).
- Angina, agravamento da angina em doentes que já sofrem de doença cardíaca.
- Ritmo cardíaco lento, dor no peito, palpitações (consciência do ritmo cardíaco), edema (acumulação de líquido), alterações no ritmo ou na velocidade dos batimentos cardíacos, insuficiência cardíaca congestiva (doença do coração com falta de ar e inchaço dos pés e pernas devido à acumulação de líquido), um tipo de perturbação do ritmo cardíaco, ataque cardíaco, insuficiência cardíaca.
- Pressão arterial baixa, fraca circulação sanguínea o que faz com que os dedos das mãos e dos pés fiquem dormentes e pálidos, mãos e pés frios.
- Falta de ar, constrição das vias aéreas nos pulmões (predominantemente em doentes com doença pré-existente), dificuldade em respirar, tosse, asma, agravamento da asma.
- Alterações do paladar, náuseas, indigestão, diarreia, boca seca, dor abdominal, vómitos.
- Queda de cabelo, erupções na pele com aparência prateada (erupção cutânea psoriasiforme) ou agravamento da psoríase, erupção na pele.
- Dor nas articulações, dor nos músculos não causada pelo exercício, fraqueza muscular, cansaço.
- Disfunção sexual, diminuição da libido.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico. Também poderá comunicar efeitos secundários diretamente através do sistema nacional de notificação mencionado abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar Monoprost Duo
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior, saqueta e recipiente unidose. O prazo de validade corresponde ao último dia do mês indicado.
Este medicamento não requer condições de temperatura especiais para conservação.
Após a primeira abertura da saqueta: utilizar os recipientes unidose no prazo de 1 mês.
Anote a data da primeira abertura na saqueta.
Após a primeira abertura do recipiente unidose: utilizar imediatamente e rejeitar o recipiente unidose após a utilização.
Manter os recipientes unidose não usados dentro da saqueta para os proteger da luz.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Monoprost Duo
As substâncias ativas são latanoprost 50 microgramas/ml e timolol (sob a forma maleato de timolol) 5mg/ml.
Os outros componentes são: hidroxistearato de macrogolglicerilo, sorbitol, macrogol, carbómero, edetato disódico, hidróxido de sódio (para ajuste do pH) e água para preparações injetáveis.
Qual o aspeto de Monoprost Duo e conteúdo da embalagem
Este medicamento apresenta-se sob a forma de colírio, solução em recipiente unidose.
A solução não tem conservantes, é ligeiramente amarelada e opalescente, praticamente isenta de partículas e está contida em recipientes unidose acondicionados numa saqueta com 5 unidades; cada recipiente unidose contém 0,2 ml de solução.
Cada embalagem contém 30 (6 x 5) ou 90 (18 x 5) recipientes unidose.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires THEA
12 rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
Fabricante
EXCELVISION
27, rue de la Lombardière
07100 Annonay
França
ou
Laboratoires THEA
12 rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Áustria, Eslováquia, França, Irlanda, Itália, Polónia, República Checa, Reino Unido…………Fixapost
Alemanha, Bélgica, Bulgária, Chipre, Espanha, Grécia, Holanda, Luxemburgo…...…………Fixaprost
Dinamarca, Estónia, Finlândia, Islândia, Lituânia, Letónia, Noruega, Suécia…………………Fixopost
Croácia, Eslovénia……………………………………………………………………………..Fixalpost
Portugal…………………………………………………………………………………Monoprost Duo
Roménia……………………………………………………………………………………….Fixanpost
Este folheto foi revisto pela última vez em 03/2022
Está disponível informação detalhada sobre este medicamento no site do INFARMED, I.P.
- Inflamação

Inflamação
Dexafree

FOLHETO INFORMATIVO: INFORMAÇÃO PARA O UTILIZADOR
DEXAFREE 1 mg/ml colírio, solução em recipiente unidose
Fosfato de dexametasona
Leia atentamente todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico. Ver secção 4.
O que contém este folheto:
1. O que é DEXAFREE e para que é utilizado
2. O que precisa de saber antes de utilizar DEXAFREE
3. Como utilizar DEXAFREE
4. Efeitos secundários possíveis
5. Como conservar DEXAFREE
6. Conteúdo da embalagem e outras informações
1. O QUE É DEXAFREE E PARA QUE É UTILIZADO
DEXAFREE é um colírio, solução que se apresenta em recipientes unidose, e contém uma substância denominada dexametasona. Esta substância pertence ao grupo dos corticosteroides, os quais inibem os sintomas inflamatórios.
DEXAFREE está indicado para o tratamento da inflamação do(s) seu(s) olho(s).
O olho não deve ter uma infeção (olho vermelho, secreções, lacrimejo…); caso contrário deverá ser associado um tratamento específico para a sua infeção (ver secção 2).
2. O QUE PRECISA DE SABER ANTES DE UTILIZAR DEXAFREE
Não utilize DEXAFREE
- se tem uma infeção ocular que pode ser de origem bacteriana (infeção purulenta aguda), fúngica, viral (herpes vírus, vírus da vaccinia, vírus da varicela zoster) ou amébica,
- se tem lesões na córnea (perfuração, ulcerações ou lesões associadas a uma cicatrização incompleta),
- se tem hipertensão ocular reconhecidamente provocada pelos glucocorticosteroides (família dos corticosteroides),
- se tem alergia (hipersensibilidade) ao fosfato sódico de dexametasona ou a qualquer outro componente de DEXAFREE.
Advertências e precauções
Fale com o seu médico ou farmacêutico antes de utilizar DEXAFREE.
NÃO INJETE, NÃO INGIRA
Evite o contacto entre a extremidade do recipiente e o olho ou as pálpebras.
- É necessária uma monitorização oftalmológica cuidadosa durante a utilização de DEXAFREE em qualquer situação, e especialmente:
- em crianças e idosos.
Recomenda-se uma monitorização oftalmológica mais frequente:
- se tem uma infeção ocular. Só deverá utilizar DEXAFREE se a infeção estiver a ser controlada com um tratamento anti-infecioso,
- se tem uma úlcera na córnea. Não utilize um tratamento tópico com dexametasona ou DEXAFREE, a não ser nos casos em que a inflamação é a principal causa do atraso na cicatrização,
- se tem uma pressão intraocular elevada. Se apresentou previamente reações adversas a um tratamento tópico com esteroides, que se traduziram num aumento da pressão intraocular, encontra-se em risco de desenvolver um aumento da pressão intraocular se for tratado com DEXAFREE.
- se tem glaucoma.
- Crianças: deverá evitar-se uma terapêutica contínua prolongada.
- Conjuntivite alérgica grave: se tem uma conjuntivite alérgica grave que não responde à terapêutica padrão, apenas deverá utilizar DEXAFREE durante um curto período de tempo.
- Diabéticos: se é diabético, deverá informar o seu oftalmologista.
- Vermelhidão no olho: se apresenta vermelhidão no olho não diagnosticada, não deverá utilizar DEXAFREE.
- Lentes de contacto: deverá evitar a utilização de lentes de contacto durante o tratamento com DEXAFREE.
Consulte o seu médico se ocorrer inchaço ou se ganhar peso à volta da cintura e no rosto, uma vez que estas são as primeiras manifestações da síndrome de Cushing. Poderá ocorrer insuficiência da função da glândula adrenal após a interrupção de um tratamento intensivo ou prolongado com o DEXAFREE. Consulte o seu médico antes de interromper o tratamento por opção própria. Estes riscos são especialmente importantes em crianças e doentes tratados com o medicamento ritonavir ou cobicistate.
Contacte o seu médico se apresentar visão turva ou outras perturbações visuais.
Outros medicamentos e DEXAFREE
No caso de um tratamento concomitante com outro colírio, solução, deve aguardar 15 minutos entre as duas aplicações.
Consulte o seu médico se estiver a tomar ritonavir ou cobicistate, uma vez que estes medicamentos podem aumentar a quantidade de dexametasona no sangue.
Têm sido referidos casos de precipitações de fosfato de cálcio na superfície da córnea quando se utilizam simultaneamente corticosteroides e bloqueadores beta tópicos.
Informe o seu médico ou farmacêutico se estiver a utilizar, ou tiver utilizado recentemente, ou se vier a utilizar outros medicamentos.
Gravidez e amamentação
- Não estão disponíveis dados suficientes relativamente à utilização de DEXAFREE durante a gravidez humana de forma a avaliar possíveis efeitos prejudiciais. Por este motivo, não se recomenda a utilização de DEXAFREE durante a gravidez.
- Não se sabe se este medicamento é excretado no leite materno. No entanto, a dose total de dexametasona é reduzida. Deste modo, DEXAFREE pode ser utilizado durante o aleitamento.
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de utilizar este medicamento.
Condução de veículos e utilização de máquinas
Tal como acontece com qualquer colírio, a turvação temporária da visão ou outras perturbações visuais, podem afetar a capacidade de conduzir ou utilizar máquinas. Não conduza ou utilize máquinas até que a visão volte ao normal.
DEXAFREE contém fosfatos
Este medicamento contém 80 microgramas de fosfatos em cada gota.
3. COMO UTILIZAR DEXAFREE
Posologia
Utilize este medicamento exatamente como indicado pelo seu médico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
A dose recomendada é de 1 gota, 4 a 6 vezes por dia no olho afetado. Em casos graves, o tratamento pode ser iniciado com 1 gota por hora e deverá ser reduzido para uma gota de 4 em 4 horas quando se observar uma resposta favorável.
Recomenda-se que a frequência de administração seja gradualmente diminuída com vista a evitar uma recaída.
- Nos idosos: não é necessário qualquer ajuste posológico.
- Nas crianças: deve evitar-se uma terapêutica contínua prolongada.
Modo de administração
Uso oftálmico: este medicamento destina-se ser aplicado no olho.
1- Lave cuidadosamente as mãos antes de utilizar o medicamento.
2- Aplique uma gota no olho afetado, olhando para cima e puxando com o dedo a pálpebra inferior para baixo.
3- Imediatamente após a aplicação, aplique uma ligeira pressão com o seu dedo na parte interna do olho tratado durante alguns minutos (para diminuir o risco de ocorrência de reações sistémicas e aumentar a penetração da substância ativa no olho).
4- Rejeite o recipiente unidose após a sua utilização. Não o conserve para uma utilização posterior.
Frequência de administração
4 a 6 vezes por dia.
Duração do tratamento
De um modo geral, a duração do tratamento varia de alguns dias até um máximo de 14 dias.
Se utilizar mais DEXAFREE do que deveria
Lave o olho com água esterilizada se instilou demasiado produto no seu olho e se apresenta uma irritação prolongada.
Contacte imediatamente o seu médico ou o seu farmacêutico.
Caso se tenha esquecido de utilizar DEXAFREE
Não utilize uma dose a dobrar para compensar uma dose que se esqueceu de utilizar.
Se parar de utilizar DEXAFREE
Não interrompa o tratamento abruptamente. Caso esteja a considerar interromper o tratamento, deverá sempre consultar o seu médico.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. EFEITOS SECUNDÁRIOS POSSÍVEIS
Como todos os medicamentos, este medicamento pode causar efeitos secundários, embora estes não se manifestem em todas as pessoas.
Doenças endócrinas:
Desconhecida: a frequência não pode ser calculada a partir dos dados disponíveis
- Problemas hormonais: crescimento de pelos corporais em excesso (especialmente nas mulheres), fraqueza ou desgaste muscular, estrias roxas, tensão arterial alta, menstruação irregular ou ausente, alterações nos níveis de proteínas e de cálcio no corpo, crescimento atrofiado em crianças e adolescentes, inchaço e ganho de peso no corpo e no rosto (Síndrome de Cushing) (consultar secção 2, “Precauções e advertências”).
Afeções oculares:
Muito frequentes: podem afetar mais de 1 em 10 pessoas
- pressão intraocular elevada, após 2 semanas de tratamento.
Frequentes: podem afetar até 1 em 10 pessoas
- desconforto, irritação, ardor, picadas, comichão e visão turva após a aplicação. Estes sintomas são normalmente temporários e ligeiros.
Pouco frequentes: podem afetar até 1 em 100 pessoas
- reações alérgicas e de hipersensibilidade a um dos componentes do colírio,
- atraso na cicatrização da ferida,
- enevoamento do cristalino do olho (catarata),
- infeções,
- glaucoma.
Muito raras: podem afetar até 1 em 10.000 pessoas
- inflamação das membranas do olho (conjuntivite),
- dilatação da pupila (midríase),
- inchaço da face (edema facial),
- pálpebras descaídas (ptose),
- inflamação da íris (uveíte),
- calcificação da córnea,
- inflamação da córnea (queratopatia do cristalino),
- alterações na espessura da córnea,
- edema da córnea,
- ulceração da córnea,
- perfuração da córnea.
Se sofrer de lesão grave na camada transparente na parte da frente do olho (córnea), os fosfatos podem causar em casos muito raros zonas nubladas na córnea devido à acumulação de cálcio durante o tratamento.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil
531749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Fax: + 351 21 798 73 97
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. COMO CONSERVAR DEXAFREE
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize DEXAFREE após o prazo de validade impresso no recipiente unidose a seguir a “Exp”.
O prazo de validade corresponde ao último dia do mês indicado.
Após a primeira abertura da saqueta: utilize os recipientes unidose no prazo de 15 dias.
Após abertura do recipiente unidose: utilize imediatamente e elimine o recipiente unidose após a sua utilização.
Manter os recipientes unidose dentro da saqueta para proteger da luz.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico.
Pergunte ao seu farmacêutico como deitar fora os medicamentos já não utiliza. Estas medidas irão ajudar a proteger o ambiente.
6. CONTEÚDO DA EMBALAGEM E OUTRAS INFORMAÇÕES
Qual a composição de DEXAFREE
- A substância ativa é o fosfato sódico de dexametasona.
Cada mililitro de solução contém 1 mg de fosfato de dexametasona (sob a forma de fosfato de sódio de dexametasona).
- Os outros componentes são edetato de dissódico, fosfato dissódico dodeca-hidratado, cloreto de sódio, água para preparações injectáveis.
Qual o aspeto de DEXAFREE e conteúdo da embalagem
DEXAFREE é uma solução límpida, incolor ou ligeiramente acastanhada, apresentada em recipientes unidose acondicionados em saquetas.
Cada recipiente unidose contém 0,4 ml de solução. As embalagens contêm 10, 20, 30, 50 ou 100 recipientes unidose.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires THEA
12, rue Louis Blériot,
63017 CLERMONT- FERRAND Cedex 2,
FRANÇA
Fabricante
EXCELVISIONR
Rue de la Lombardière,
07100 Annonay,
FRANÇA
Ou
Laboratoire UNITHER
1 rue de l’Arquerie
50200 COUTANCES
FRANÇA
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Espanha, França, Polónia e Portugal-DEXAFREE
Bélgica, Holanda e Luxemburgo-MONOFREE DEXAMETHASON
Alemanha e Áustria-MONODEX
Itália-DEXAMONO
Este folheto foi revisto pela última vez em: 11/2021

Inflamação
Voltaren
Folheto informativo: Informação para o utilizador
Voltaren 1 mg/ml Colírio, solução
Diclofenac sódico
Leia com atenção todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico ou farmacêutico. Ver secção 4.
O que contém este folheto:
1. O que é Voltaren e para que é utilizado
2. O que precisa de saber antes de utilizar Voltaren
3. Como utilizar Voltaren
4. Efeitos indesejáveis possíveis
5. Como conservar Voltaren
6. Conteúdo da embalagem e outras informações
1. O que é Voltaren e para que é utilizado
Voltaren é um colírio, solução embalado em frascos de 5 ml.
O diclofenac sódico 1 mg/ml, substância ativa do Voltaren, tem propriedades anti-inflamatórias e analgésicas, e destina-se a ser aplicado no globo ocular, como tratamento local.
Voltaren tem as seguintes indicações terapêuticas:
- Inflamação pós-operatória na cirurgia de cataratas e outras intervenções cirúrgicas;
- profilaxia pré e pós-operatória do edema macular cistoide associado a extração de cataratas e implantação de lente intraocular;
- Inflamação pós-traumática em feridas não penetrantes;
- Alívio da dor ocular e fotofobia após cirurgia refrativa da córnea.
2. O que precisa de saber antes de utilizar Voltaren
O frasco mantêm-se estéril até violação do anel de segurança. Não toque com a extremidade conta-gotas no olho ou em qualquer outra superfície, para evitar a contaminação da solução.
Não utilize Voltaren:
- se tem alergia ao diclofenac sódico ou a qualquer outro componente deste medicamento (indicados na secção 6).
- se sofre de asma, urticária ou rinite aguda, precipitáveis pelo ácido acetilsalicílico ou por outros fármacos inibidores da síntese das prostaglandinas.
Voltaren colírio não deve ser utilizado por via intra-ocular durante a cirurgia.
Advertências e precauções
Fale com o seu médico ou farmacêutico antes de utilizar Voltaren.
Devido à possibilidade meramente teórica de absorção da substância activa de Voltaren é necessária precaução na utilização deste medicamento em idosos, crianças e adolescentes, grávidas em início de gravidez e durante o aleitamento, doentes com antecedentes de doenças gástricas ou hemorragias digestivas, doenças de rins e doentes a tomar anticoagulantes orais.
Outros medicamentos e Voltaren
Informe o seu médico ou farmacêutico se estiver a tomar, ou tiver tomado recentemente, ou se vier a tomar outros medicamentos.
Se utiliza outros colírios, deve esperar 5 minutos entre as aplicações.
Gravidez e Amamentação
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento.
Gravidez
Não utilize Voltarense estiver nos últimos 3 meses de gravidez. Não deve utilizar Voltarendurante os primeiros 6 meses de gravidez, a menos que seja claramente necessário e aconselhado pelo seu médico. Se necessitar de tratamento durante este período, deverá ser utilizada a dose mais baixa e pelo menor tempo possível.
As formas orais (por exemplo, comprimidos) de diclofenacpodem causar efeitos adversos no feto em gestação. Não se sabe se o mesmo risco se aplica aVoltarenquando é utilizado no olho.
Amamentação
Após a administração oral de comprimidos revestidos de 50 mg (o conteúdo de 10 frascos de Voltaren colírio), apenas foram detetados vestígios de substância ativa no leite materno e em quantidades tão baixas que não são esperadas reações adversas na criança. Não se recomenda o uso ocular de Voltaren colírio durante o aleitamento, a não ser que o seu médico dê informações em contrário para o seu caso.
Condução de veículos e utilização de máquinas
Se a sua visão estiver enevoada evite conduzir veículos ou usar máquinas.
Voltaren contém cloreto de benzalcónio
Este medicamento contém 0,0014 mg de cloreto de benzalcónio em cada gota que é equivalente a 0,05 mg/ml.
O cloreto de benzalcónio pode ser absorvido pelas lentes de contacto moles e pode alterar a cor das lentes de contacto. Deve remover as lentes de contacto antes da utilização deste medicamento e voltar a colocá-las 15 minutos depois.
O cloreto de benzalcónio pode também causar irritação nos olhos, especialmente se tem os olhos secos ou alterações da córnea (a camada transparente na parte da frente do olho). Se tiver uma sensação estranha no olho, picadas ou dor no olho após utilizar este medicamento, fale com o seu médico.
3. Como utilizar Voltaren
Utilize este medicamento exatamente como indicado pelo seu médico ou farmacêutico.
Fale com o seu médico ou farmacêutico se tiver dúvidas.
Voltaren pode ser utilizado em adultos e idosos. A experiência em crianças com mais de 2 anos na cirurgia do estrabismo é limitada.
Voltaren é um medicamento para uso oftálmico.
A dosagem é a seguinte:
Na cirurgia ocular e suas complicações:
- pré-operatório: 1 gota até 5 vezes, nas 3 horas antes da cirurgia.
- pós-operatório: 1 gota 3 vezes no dia da cirurgia, seguindo-se 1 gota, 3 a 5 vezes por dia, durante tanto tempo quanto o necessário.
Alívio da dor ocular e fotofobia; inflamação pós-traumática:
- 1 gota de 4 em 4 ou de 6 em 6 horas.
Para aplicar o medicamento, lave primeiro as mãos, puxe a pálpebra inferior ligeiramente para baixo com uma mão e segure-a. Incline a sua cabeça para trás. Com a outra mão, segure o frasco virado para baixo por cima do olho a ser tratado. Com cuidado, aperte-o até que caia uma gota no seu olho.
Fale com o seu médico ou farmacêutico se tiver a impressão de que Voltaren é demasiado forte ou demasiado fraco.
Se utilizar mais Voltaren do que deveria
Não há perigo em ingerir Voltaren acidentalmente. Da mesma forma, não se preocupe se cair mais do que uma gota no seu olho. Se tiver dúvidas, aconselhe-se junto do seu médico ou farmacêutico.
Caso se tenha esquecido de utilizar Voltaren
Se se tiver esquecido de administrar uma dose, aplique essa dose o mais cedo possível e retome o seu horário de dosagem regular. Certifique-se que a gota caiu no olho.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos indesejáveis possíveis
Como todos os medicamentos, este medicamento pode causar efeitos indesejáveis, embora estes não se manifestem em todas as pessoas.
A reação adversa mais frequentemente observada foi sensação de ardor nos olhos, transitória, ligeira a moderada. Muito frequentemente foram notificados casos de dor nos olhos. Outras reações menos frequentemente observadas foram comichão, vermelhidão e visão enevoada imediatamente após aplicação do colírio. Foram observadas queratites ponteadas ou disfunções da córnea (usualmente após aplicações frequentes).
Em doentes com factores de risco para o aparecimento de disfunções da córnea, tais como durante a utilização de corticosteróides ou com doenças concomitantes como infeções ou artrite reumatóide, o diclofenac foi associado, em casos raros, com queratite ulcerosa, adelgaçamento corneano, queratite ponteada, alterações no epitélio da córnea e edema da córnea, que pode colocar a visão em risco. A maioria dos doentes tinham sido tratados por longos períodos de tempo.
Em casos raros, foi observada dificuldade em respirar e agravamento da asma.
Foram notificadas situações alérgicas, como por exemplo, hiperemia conjuntival, conjuntivite alérgica, eritema das pálpebras, alergia ocular, edema das pálpebras, prurido nas pálpebras, urticária, erupção cutânea, eritema, prurido, hipersensibilidade, tosse e rinite.
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos indesejáveis diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos indesejáveis, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
(preferencialmente)
ou através dos seguintes contactos:
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar Voltaren
Não conservar acima de 25ºC.
Após a abertura usar no prazo de 4 semanas.
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior, após VAL. O prazo de validade corresponde ao último dia do mês indicado.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Voltaren
- A substância ativa é o diclofenac sódico. Cada militro de colírio, solução contém 1 mg de diclofenac sódico.
- Os outros componentes são: cloreto de benzalcónio, edetato dissódico, hidroxipropilgama-ciclodextrina, ácido clorídrico, propilenoglicol (E1520), trometamol, tiloxapol e água para preparações injetáveis.
Qual o aspeto de Voltaren e conteúdo da embalagem
Voltaren apresenta-se como uma solução transparente e sem odor.
Voltaren é um colírio, solução acondicionado em frasco conta-gotas com capacidade de 5 ml e tampa HDPE.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires THEA
12, rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
Fabricante
Excelvision
Rue de la Lombardière
07100 Annonay
França
Este folheto foi revisto pela última vez em 10/2024
Folheto informativo: Informação para o utilizador
Voltaren Colírio Unidoses 0,3 mg/0,3 ml Colírio, solução
Diclofenac sódico
Leia com atenção todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico ou farmacêutico. Ver secção 4.
O que contém este folheto:
1. O que é Voltaren Colírio Unidoses e para que é utilizado
2. O que precisa de saber antes de utilizar Voltaren Colírio Unidoses
3. Como utilizar Voltaren Colírio Unidoses
4. Efeitos indesejáveis possíveis
5. Como conservar Voltaren Colírio Unidoses
6. Conteúdo da embalagem e outras informações
1. O que é Voltaren Colírio Unidoses e para que é utilizado
Voltaren Colírio Unidoses é um colírio ocular estéril em unidoses, sem conservantes. Cada unidose contém 0,3 ml de colírio,solução.
O diclofenac sódico 1 mg/ml, substância ativa do Voltaren Colírio Unidoses, tem propriedades anti-inflamatórias e analgésicas, e destina-se a ser aplicado no globo ocular, como tratamento local.
Voltaren Colírio Unidoses tem as seguintes indicações terapêuticas:
- Inflamação pós-operatória da cirurgia de cataratas e outras intervenções cirúrgicas.
- Profilaxia pré e pós-operatória do edema macular cistoide associado a extração de cataratas e implantação de lente intraocular;
- Inflamação pós-traumática em feridas não-penetrantes;
- Alívio da dor ocular e fotofobia após cirurgia refrativa da córnea.
2. O que precisa de saber antes de utilizar Voltaren Colírio Unidoses
Não utilize Voltaren Colírio Unidoses:
- se tem alergia ao diclofenac sódico ou a qualquer outro componente deste medicamento (indicados na secção 6).
- se sofre de asma, urticária ou rinite aguda, precipitáveis pelo ácido acetilsalicílico ou por outros fármacos inibidores da síntese das prostaglandinas.
Voltaren colírio não deve ser utilizado por via intraocular durante a cirurgia.
Advertências e precauções
Fale com o seu médico ou farmacêutico antes de utilizar Voltaren Colírio Unidoses.
Devido à possibilidade meramente teórica de absorção da substância ativa de Voltaren Colírio Unidoses é necessária precaução na utilização deste medicamento em idosos, crianças e adolescentes, grávidas em início de gravidez e durante o aleitamento, doentes com antecedentes de doenças gástricas ou hemorragias digestivas, doenças de rins e doentes a tomar anticoagulantes orais.
Outros medicamentos e Voltaren Colírio Unidoses
Informe o seu médico ou farmacêutico se estiver a tomar, ou tiver tomado recentemente, ou se vier a tomar outros medicamentos.
Se utiliza outros colírios, deve esperar 5 minutos entre as aplicações.
Gravidez e Amamentação
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento.
Gravidez
Não utilize Voltaren Colírio Unidoses se estiver nos últimos 3 meses de gravidez. Não deve utilizar Voltaren Colírio Unidoses durante os primeiros 6 meses de gravidez, a menos que seja claramente necessário e aconselhado pelo seu médico. Se necessitar de tratamento durante este período, deverá ser utilizada a dose mais baixa e pelo menor tempo possível.
As formas orais (por exemplo, comprimidos) de diclofenac podem causar efeitos adversos no feto em gestação. Não se sabe se o mesmo risco se aplica a Voltaren Colírio Unidoses quando é utilizado no olho.
Amamentação
Após a administração oral de comprimidos revestidos de 50 mg (o conteúdo de 10 frascos de Voltaren, colírio, solução a 1 mg/ml), apenas foram detetados vestígios de substância ativa no leite materno e em quantidades tão baixas que não são esperadas reações adversas na criança.
Condução de veículos e utilização de máquinas
Se a sua visão estiver enevoada evite conduzir veículos ou usar máquinas.
3. Como utilizar Voltaren Colírio Unidoses
Utilize este medicamento exatamente como indicado pelo seu médico ou farmacêutico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
Voltaren Colírio Unidoses pode ser utilizado em adultos e idosos. A experiência em crianças com mais de 2 anos na cirurgia do estrabismo é limitada.
Voltaren Colírio Unidoses é um medicamento para uso oftálmico.
A dosagem é a seguinte:
Na cirurgia ocular e suas complicações:
- pré-operatório: 1 gota, até 5 vezes, nas 3 horas antes da cirurgia;
- pós-operatório: 1 gota,, 3 vezes no dia da cirurgia, seguindo-se 1 gota, 3 a 5 vezes por dia, durante tanto tempo quanto necessário.
Alívio da dor ocular e fotofobia; inflamação pós-traumática:
- 1 gota de 4 em 4 ou de 6 em 6 horas.
Para aplicar o medicamento, lave primeiro as mãos, puxe a pálpebra inferior ligeiramente para baixo com uma mão e segure-a. Incline a sua cabeça para trás. Com a outra mão, segure o frasco virado para baixo por cima do olho a ser tratado. Com cuidado, aperte-o até que caia uma gota no seu olho.
Fale com o seu médico ou farmacêutico se tiver a impressão de que Voltaren Colírio Unidoses é demasiado forte ou demasiado fraco.
Nota: rejeitar a unidose após utilização, mesmo que não tenha usado a totalidade do volume. Nunca guarde o volume remanescente para uso posterior, uma vez que a solução não tem conservantes.
Se utilizar mais Voltaren Colírio Unidoses do que deveria
Não há perigo em ingerir Voltaren Colírio Unidoses, acidentalmente. Da mesma forma, não se preocupe se cair mais do que uma gota no seu olho. Se tiver dúvidas, aconselhe-se junto do seu médico ou farmacêutico.
Caso se tenha esquecido de utilizar Voltaren Colírio Unidoses
Se se tiver esquecido de administrar uma dose, aplique essa dose o mais cedo possível e retome o seu horário de dosagem regular. Certifique-se que a gota caiu no olho.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico ou farmacêutico.
4. Efeitos indesejáveis possíveis
Como todos os medicamentos, este medicamento pode causar efeitos indesejáveis, embora estes não se manifestem em todas as pessoas.
A reação adversa mais frequentemente observada foi sensação de ardor nos olhos, transitória, ligeira a moderada. Muito frequentemente foram notificados casos de dor nos olhos. Outras reações menos frequentemente observadas foram, comichão, vermelhidão e visão enevoada imediatamente após aplicação do colírio. Foram observadas queratites ponteadas ou disfunções da córnea (usualmente após aplicações frequentes).
Em doentes com fatores de risco para o aparecimento de disfunções da córnea, tais como durante a utilização de corticosteroides ou com doenças concomitantes como infeções ou artrite reumatoide, o diclofenac sódico foi associado, em casos raros, com queratite ulcerosa, adelgaçamento da córnea, queratite ponteada, alterações no epitélio da córnea e edema da córnea que pode colocar a visão em risco. A maioria dos doentes tinham sido tratados por longos períodos de tempo.
Em casos raros, foi observada dificuldade em respirar e agravamento da asma.
Foram notificadas situações alérgicas, como por exemplo, hiperemia conjuntival, conjuntivite alérgica, eritema das pálpebras, alergia ocular, edema das pálpebras, prurido nas pálpebras, urticária, erupção cutânea, eritema, prurido, hipersensibilidade, tosse e rinite.
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos indesejáveis diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos indesejáveis, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
(preferencialmente)
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar Voltaren Colírio Unidoses
Não conservar acima de 25ºC.
Após a primeira abertura do blister: o medicamento é estável durante 4 semanas quando conservado a temperatura inferior a 25ºC.
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior, após VAL. O prazo de validade corresponde ao último dia do mês indicado.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de Voltaren Colírio Unidoses
- A substância ativa é o diclofenac sódico. Cada mililitro de Voltaren Colírio Unidoses contém 1 mg de diclofenac sódico.
- Os outros componentes são: ácido bórico, trometamol, óleo de rícino de polietilenoglicol 35 e água para preparações injetáveis.
Qual o aspeto de Voltaren Colírio Unidoses e conteúdo da embalagem
Voltaren Colírio Unidoses apresenta-se como uma solução ligeiramente amarelada e opaca, com odor característico.
Embalagens contendo 10, 20 ou 40 recipientes unidose transparentes (blocos de 5 unidoses cada), em polietileno de baixa densidade. Cada bloco de 5 unidoses está acondicionado em blister de alumínio/PVC.
Cada recipiente unidose contém 0,3 ml.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires THEA
12, rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
Fabricante
Excelvision
Rue de la Lombardière
07100 Annonay
França
Este folheto foi revisto pela última vez em 10/2024

Inflamação
Softacort
Folheto informativo: Informação para o doente
Softacort 3,35 mg/ml colírio, solução em recipiente unidose
Fosfato sódico de hidrocortisona
Leia com atenção todo este folheto antes de começar a utilizar este medicamento, pois contém informação importante para si.
- Conserve este folheto. Pode ter necessidade de o ler novamente.
- Caso ainda tenha dúvidas, fale com o seu médico, farmacêutico ou enfermeiro.
- Este medicamento foi receitado apenas para si. Não deve dá-lo a outros. O medicamento pode ser-lhes prejudicial mesmo que apresentem os mesmos sinais de doença.
- Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Ver secção 4.
O que contém este folheto:
- O que é Softacort e para que é utilizado
- O que precisa de saber antes de utilizar Softacort
- Como utilizar Softacort
- Efeitos idesejáveis possíveis
- Como conservar Softacort
- Conteúdo da embalagem e outras informações
1. O que é Softacort e para que é utilizado
Este medicamento é um colírio, solução em recipiente unidose o qual contém uma substância denominada hidrocortisona. Esta substância é um corticosteroide que inibe os sintomas inflamatórios.
É utilizado para tratar situações inflamatórias ou alérgicas ligeiras da parte superficial do(s) seu(s) olho(s) (conjuntiva).
O olho não deve estar infetado (ver “Não utilize Softacort”).
2 O que precisa de saber antes de utilizar Softacort
Não utilize Softacort:
- Se tem alergia à substância ativa (hidrocortsiona) ou a qualquer outro componente deste medicamento (indicados na secção 6).
- Se tem uma pressão intraocular elevada (hipertensão ocular) que se sabe ser devida aos glucocorticoides (família dos corticosteroides) ou ter outras causas.
- Se tem uma infeção viral aguda por herpes e a maioria de outras infeções virais na fase de ulceração (a não ser que a infeção esteja a ser tratada com um tratamento anti-infecioso para o vírus do herpes).
- Se tem uma conjuntivite com inflamação ulcerativa da córnea (queratite) mesmo na fase inicial.
- Se tem uma infeção bacteriana no olho (infeção purulenta aguda, conjuntivite, blefarite ou terçolho).
- Se tem uma infeção fúngica no olho (micose ocular).
- Se tem uma infeção bacteriana denominada tuberculose que afeta o seu olho (tuberculose ocular).
Advertências e precauções
- Fale com o seu médico, farmacêutico ou enfermeiro antes de utilizar este medicamento.
- Se tem um olho vermelho que não foi diagnosticado, não utilize este medicamento.
- Se tem um infeção ocular viral (herpes), utilize apenas este medicamento caso a infeção esteja a ser tratada com um tratamento anti-infecioso e será necessária uma monitorização cuidadosa dos seus olhos.
- Se tem uma doença que causa uma diminuição da espessura da parte exterior do seu olho (córnea e esclerótida), poderá haver um risco aumentado de perfuração devido à utilização de corticosteroides tópicos aplicados no olho.
- Se utiliza ou utilizou um corticosteroide durante um longo período de tempo e se tem uma ferida no olho (úlcera na córnea), poderá suspeitar-se de uma infeção fúngica.
- Será necessária uma monitorização cuidadosa regular dos seus olhos durante o seu tratamento. Uma utilização prolongada de corticosteroides demonstrou causar um aumento da pressão no interior do olho e o desencadeamento de glaucoma, especialmente em doentes que já sofrem de pressão intraocular elevada ou em doentes em risco de desenvolver tal condição com um tratamento esteroide local (ver “Efeitos indesejáveis possíveis”) e pode provocar um enevoamento do cristalino no olho (catarata), em particular em crianças e na população idosa.
- A utilização de corticosteroides pode causar infeções oculares oportunistas. Além disso, os corticosteroides oculares tópicos podem causar, agravar ou mascarar os sinais e sintomas das infeções oculares oportunistas.
- Deve evitar utilizar lentes de contacto durante o tratamento com este medicamento.
- Contacte o seu médico se apresentar visão turva ou outras perturbações visuais.
Crianças
Não existem dados de segurança e eficácia em crianças.
O tratamento contínuo e prolongado com corticosteroides em crianças pode causar supressão suprarrenal.
O aumento da pressão ocular em crianças ocorre mais frequentemente, com maior gravidade e mais rapidamente que nos adultos.
Outros medicamentos e Softacort
Informe o seu médico ou farmacêutico se estiver a utilizar, tiver utilizado recentemente, ou se vier a utilizar outros medicamentos, incluindo medicamentos obtidos sem receita médica.
Alguns medicamentos podem aumentar os efeitos de Softacort e o seu médico poderá querer monitoriza-lo cuidadosamente caso esteja a tomar estes medicamentos (incluindo alguns medicamentos para o VIH: ritonavir, cobicistato).
Gravidez e amamentação
Se está grávida ou a amamentar, se pensa estar grávida ou planeia engravidar, consulte o seu médico ou farmacêutico antes de tomar este medicamento.
- A utilização deste medicamento durante a gravidez não é recomendada exceto quando o seu médico considerar necessário e sob rigorosa supervisão.
- Não se sabe se este medicamento passa para o leite materno. O seu médico irá decidir se pode utilizar ou não este medicamento durante a amamentação.
Condução de veículos e utilização de máquinas
Visão turva temporária ou outras perturbações visuais podem afetar a capacidade de conduzir ou utilizar máquinas. Não conduza ou utilize máquinas até que a sua visão volte ao normal.
Softacort contém fosfatos
Este medicamento contém 0,227 mg de fosfatos em cada gota.
3 Como utilizar Softacort
Dose
Utilize este medicamento exatamente como indicado pelo seu médico ou farmacêutico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
A dose recomendada é de 2 gotas no(s) olho(s) afetado(s), 2 a 4 vezes por dia, dependendo da prescrição.
É recomendado efetuar uma redução gradual da dose para evitar recidivas. A duração do tratamento normalmente varia de alguns dias até um máximo de 14 dias.
A mesma dose é utilizada para adultos e idosos.
Utilização em crianças
A eficácia e segurança em crianças não foram estabelecidas.
Como utilizar o colírio
Este medicamento destina-se a ser administrado no olho.
Siga, por favor, as seguintes instruções para utilizar o colírio:
1. Lave as suas mãos e sente-se ou coloque-se numa posição confortável.
2. Abra a saqueta que contém 10 recipientes unidose. Escreva a data da primeira abertura na saqueta.
3. Retire um recipiente unidose da tira.
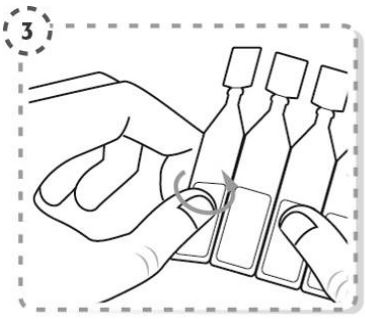
4. Rode a extremidade superior do recipiente unidose tal como indicado na figura. Não toque na ponta após a abertura do recipiente.
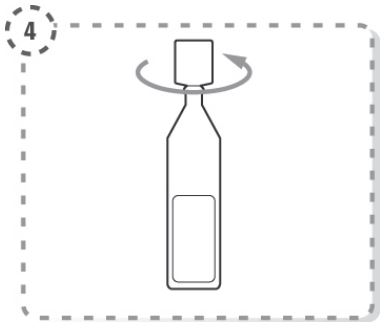
5. Utilize o seu dedo para puxar cuidadosamente para baixo a pálpebra inferior do olho afetado.
6. Coloque a extremidade do recipiente unidose próximo do seu olho, mas de modo a não lhe tocar.
7. Pressione cuidadosamente o recipiente unidose de forma que entrem duas gotas no seu olho, depois liberte a pálpebra inferior.
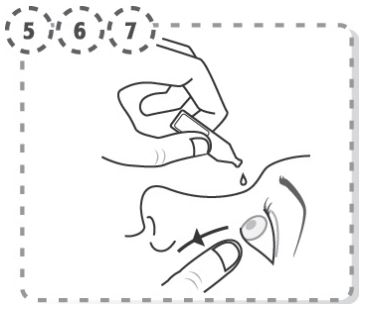
8. Com um dedo pressione o canto do olho, mais próximo do nariz. Pressione durante 1 minuto, enquanto mantém o olho fechado.
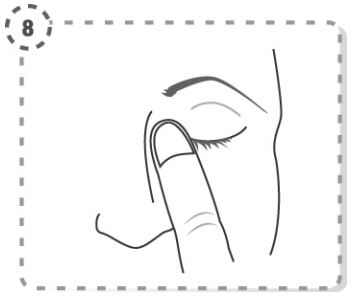
9. Repita esta operação no outro olho, se o seu médico lhe tiver dito para o fazer. Cada recipiente unidose contém solução suficiente para ambos os olhos.
10. Rejeite o recipiente unidose após a utilização. Não conserve o recipiente para uma utilização posterior.
11. Coloque os recipientes unidose que não foram abertos dentro da saqueta. Coloque a saqueta aberta dentro da embalagem. Os recipientes unidose que não foram abertos têm de ser utilizados no espaço de um mês após a abertura da saqueta.
Caso esteja a utilizar outro medicamento de aplicação ocular, deve aguardar 5 minutos entre cada aplicação.
Se tomar utilizar mais Softacort do que deveria
Enxague o olho com água estéril se tiver aplicado demasiado produto no seu olho e se tiver uma irritação prolongada.
Contacte imediatamente o seu médico ou farmacêutico.
Caso se tenha esquecido de utilizar Softacort
Não utilize uma dose a dobrar para compensar uma dose que se esqueceu de utilizar.
Se parar de utilizar Softacort
Não pare de utilizar o tratamento abruptamente. Consulte sempre o seu médico caso esteja a considerar interromper o tratamento.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico, farmacêutico ou enfermeiro.
4 Efeitos idesejáveis possíveis
Como todos os medicamentos, este medicamento pode causar efeitos indesejáveis, embora estes não se manifestem em todas as pessoas.
Desconhecida: a frequência não pode ser calculada a partir dos dados disponíveis
- Desconforto ocular temporário (sensação de queimadura ou picadas) após a aplicação.
Os seguintes efeitos indesejáveis foram notificados com medicamentos do mesmo grupo (corticosteroides) quando utilizados no tratamento de doenças oculares.
Desconhecida: a frequência não pode ser calculada a partir dos dados disponíveis
- reações alérgicas,
- atraso na cicatrização de feridas,
- enevoamento do cristalino no olho (catarata capsular posterior),
- infeções oportunistas (infeção fúngica e viral do tipo herpes),
- aumento da pressão no seu olho (glaucoma),
- dilatação da pupila (midríase),
- descaimento das pálpebras (ptose),
- inflamação no interior do seu olho (uveíte),
- alterações na espessura da camada exterior do olho (córnea),
- inflamação da córnea (queratopatia do cristalino),
- visão turva.
Se sofrer de lesão grave na camada transparente na parte da frente do olho (córnea), os fosfatos podem causar em casos muito raros zonas nubladas na córnea devido à acumulação de cálcio durante o tratamento.
Comunicação de efeitos indesejáveis
Se tiver quaisquer efeitos indesejáveis, incluindo possíveis efeitos indesejáveis não indicados neste folheto, fale com o seu médico, farmacêutico ou enfermeiro. Também poderá comunicar efeitos indesejáveis diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos indesejáveis, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
Sítio da internet:
http://www.infarmed.pt/web/infarmed/submissaoram (preferencialmente)
ou através dos seguintes contactos:
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
E-mail: farmacovigilancia@infarmed.pt
5 Como conservar Softacort
Manter este medicamento fora da vista e do alcance das crianças.
Não utilize este medicamento após o prazo de validade impresso na embalagem exterior, saqueta e recipiente unidose após EXP. O prazo de validade corresponde ao último dia do mês indicado.
Não conservar acima de 25ºC.
Após a primeira abertura da saqueta: utilize os recipientes unidose no prazo de um mês.
Mantenha os recipientes unidose dentro da saqueta para os proteger da luz.
Escreva a data da primeira abertura na saqueta.
Após a primeira abertura do recipiente unidose: utilize imediatamente e elimine o recipiente unidose após a sua utilização.
Uma vez que a esterilidade não pode ser mantida após a abertura do recipiente unidose individual, tem de abrir um novo recipiente unidose antes de cada utilização.
Não deite fora quaisquer medicamentos na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como deitar fora os medicamentos que já não utiliza. Estas medidas ajudarão a proteger o ambiente.
6 Conteúdo da embalagem e outras informações
Qual a composição de Softacort
- A substância ativa é o fosfato sódico de hidrocortisona.
1 ml de colírio, solução contém 3,35 mg de fosfato sódico de hidrocortisona.
- Os outros componentes são fosfato dissódico dodeca-hidratado, di-hidrogenofosfato de sódio mono-hidratado, cloreto de sódio, edetato dissódico, ácido clorídrico (para ajuste do pH), água para preparações injetáveis.
Qual o aspeto de Softacort e conteúdo da embalagem
Este medicamento apresenta-se sob a forma de colírio, solução em recipiente unidose.
A solução é praticamente límpida, incolor ou ligeiramente amarelada, praticamente isenta de partículas. É acondicionada em saquetas contendo 10 unidades e cada recipiente unidose contém 0,4 ml de produto.
As embalagens contêm 10 (1x10), 20 (2x10), 30 (3x10) ou 60 (6x10) recipientes unidose.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
LABORATOIRES THEA
12, rue Louis Blériot
63017 CLERMONT-FERRAND Cedex 2
FRANÇA
Fabricante
LABORATOIRES UNITHER
1 rue de l’Arquerie
50200 Coutances
FRANÇA
ou
LABORATOIRES THEA
12, rue Louis Blériot
63017 CLERMONT-FERRAND Cedex 2
FRANÇA
Este medicamento encontra-se autorizado nos Estados Membros do Espaço Económico Europeu (EEE) sob as seguintes denominações:
Alemanha, Áustria, Bulgária, Chipre, Croácia, Dinamarca, Eslováquia, Eslovénia, Espanha, Grécia, Finlândia, França, Islândia, Noruega, Polónia, Portugal, Reino Unido, República Checa, Roménia, Suécia - Softacort
Bélgica, Holanda, Luxemburgo - Softacor
Irlanda - Zoftacot
Itália - Sofacor
Este folheto foi revisto pela última vez em 11/2023
- Nutrição

Nutrição
Meralut

MERALUT®
Suplemento alimentar
Suplemento de vitamina A, fl avonoides naturais, luteína e zeaxantina
|
Informação nutricional |
||||
| por 1 cápsula | %VRN | por 2 cápsulas | %VRN* | |
| Vitaminas | ||||
| Vitamina A (retinol) | 400 µg RE | 50% | 800 µg | 100% |
| Outras substâncias | ||||
| Flavonoides | 50 mg | 100 mg | ||
| Luteína | 1,5 mg | 3 mg | ||
| Zeaxantina | 0,075 mg | 0,15 mg | ||
| * VRN: valores de referência do nutriente | ||||
| Valor energético | ||||
| por 1 cápsula | Por 2 cápsulas | |||
| Kcal | 4 | 8 | ||
| KJ | 15 | 30 | ||
Ingredientes:
Óleo de girassol, emulsionante (monoestearato de glicerilo), gelatina**, glicerol**, mirtilo desidratado (flavonoides 25%), extrato natural rico em luteína (20%) e zeaxantina (1%), palmitato de retinol e corantes** (E133 e E172).
** ingredientes utilizados no revestimento da cápsula
MERALUT® foi desenvolvido para complementar as necessidades diárias de vitamina A, com um aporte adicional de flavonoides naturais, luteína e zeaxantina.
O aporte suficiente destes nutrientes e substâncias através da alimentação, complementando-a quando necessário, favorece o correto funcionamento da retina e a saúde ocular.
Não exceder a dose diária recomendada.
Os suplementos alimentares não devem ser utilizados em substituição de um regime alimentar equilibrado
Modo de utilização
Recomendam-se duas cápsulas por dia tomadas com um copo de água durante as refeições.
Conservação
Conservar em local seco e fresco.
Não consumir após a data de validade indicada na embalagem.
Apresentação
Embalagem contendo 21 g (30 cápsulas).
MANTER FORA DO ALCANCE DAS CRIANÇAS.
N7090CA30LTE/0915 - 200058

Nutrição
Nutrof Omega

NUTROF® OMEGA
NUTROF OMEGA é um suplemento alimentar à base de DHA, EPA, Zn, Mn, Se, Cu, Vitaminas C, D e E, Complexo B, Luteina e Zeaxantina.
Administração oral.
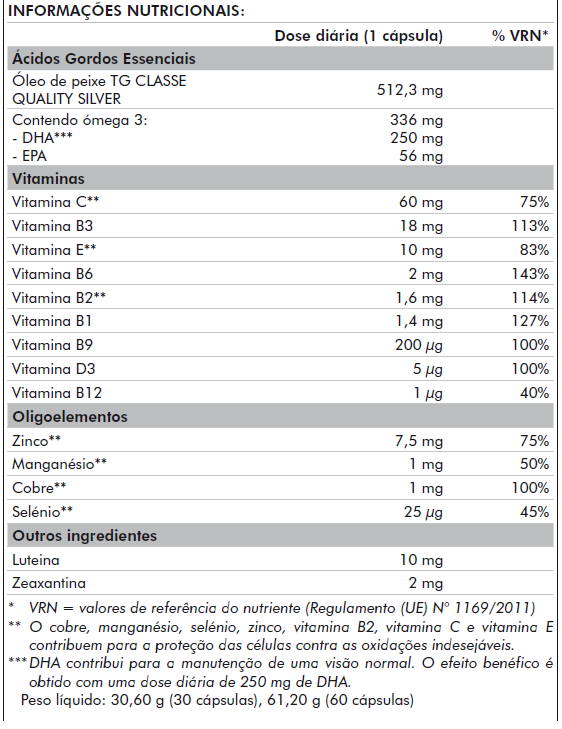
COMPOSIÇÃO:
Óleo de peixe (ácidos gordos essenciais ómega 3), gelatina, água purificada, vitamina C (ácido L-ascórbico), luteína, óleo de cártamo, humidificante: glicerol, sorbitol, emulsificante: monoestearato de glicerol, vitamina B3 (nicotinamida), vitamina E (dl-alfa tocoferol acetato), zeaxantina, óleo de cártamo, Emulsificante: lecitina de soja, zinco (óxido), vitamina B6 (cloridrato de piridoxina), manganésio (sulfato), cobre (sulfato), vitamina B1 (nitrato de tiamina), vitamina B2 (riboflavina), vitamina B12 (cianocobalamina 0,1%), glutationa, corante: óxido negro de ferro, óxido de ferro vermelho, vitamina B9 (ácido fólico), vitamina D (colecalciferol), selénio (selenito de sódio).
MODO DE UTILIZAÇÃO:
NUTROF OMEGA foi estudado para completar as necessidades diárias de: DHA, EPA, Vitaminas C, D e E e do grupo B, oligoelementos essenciais (Zinco, Manganésio, Selénio e Cobre), Luteina e Zeaxantina.
Recomenda-se a toma de 1 cápsula por dia, acompanhada de um copo de água à refeição.
Não exceder a dose diária recomendada. Os suplementos alimentares não devem substituir uma dieta equilibrada e variada nem um modo de vida saudável.
CONSERVAÇÃO:
Conservar a uma temperatura inferior a 25ºC, ao abrigo da luz e da humidade.
A cápsula de gelatina pode absorver a humidade.
MANTER FORA DO ALCANCE DAS CRIANÇAS.
PRAZO DE VALIDADE:
Não utilizar após o prazo de validade indicado na embalagem.
Laboratoires Théa
12, rue Louis Blériot
63017 Clermont-Ferrand
Cedex 2

Nutrição
Hyabak Cáps

Hyabak Caps®
Ácidos gordos essenciais; vitaminas e oligoelementos antioxidantes* para a manutenção de uma visão normal.
HYABAK CAPS® é um suplemento alimentar que contém ácidos gordos essenciais ómega 3 e ómega 6, vitaminas e oligoelementos antioxidantes* para a manutenção de uma visão normal.
O zinco contribui para a manutenção de uma visão normal.
| INFORMAÇÃO NUTRICIONAL | ||
| Dose Diária (2 cápsulas) | % VRN ** | |
| Ácidos gordos essenciais | ||
| Óleo de peixe do qual 70 % DHA (Omega 3) | 560 mg | |
| Óleo de borragem do qual 22 % GLA (Omega 6) | 420 mg | |
| Vitaminas | ||
| Vitamina C | 80 mg | 100 % |
| Vitamina E | 10 mg | 83 % |
| Vitamina B6 | 2 mg | 143 % |
| Vitamina B9 | 200 µg | 100 % |
| Vitamina B12 | 1 µg | 40 % |
| Minerais | ||
| Zinco | 10 mg | 100 % |
|
* O zinco, a vitamina E e vitamina C contribuem para a proteção das células contra as oxidações indesejáveis. ** VRN = valores de referência do nutriente (Regulamento (UE) N° 1169/2011) |
||
INGREDIENTES QUALITYSILVER®
óleo de peixe (DHA 70 %), óleo de borragem (Borago officinalis) (GLA 22 %), gelatina (agente de revestimento), água purificada, vitamina C (ácido ascórbico), glicerol (espessante), sorbitol (humidificante), monoestearato de glicerol (emulsionante), cera de abelhas (antiaglomerante), óleo de soja (emulsionante), lecitina de soja (emulsionante), vitamina E (acetato de DL-alfatocoferilo), zinco (óxido), vitamina B6 (cloridrato de piridoxina), óxido de ferro negro (corante), vitamina B12 (cianocobalamina 0,1 %), óxido de ferro vermelho (corante), vitamina B9 (ácido fólico).
MODO DE UTILIZAÇÃO
1 cápsula duas vezes por dia: Recomenda-se a toma das cápsulas durante a refeição e com um pouco de água.
HYABAK CAPS® é um suplemento alimentar e não deve substituir um regime alimentar equilibrado e variado nem um estilo de vida saudável.
Não ultrapassar a dose diária recomendada.
CONSERVAÇÃO
- Manter fora do alcance das crianças.
- Conservar a temperatura inferior a 25°C, ao abrigo da luz e da humidade.
COMERCIALIZADO POR
Théa Portugal S.A.
Rua Pedro Álvares de Cabral
Edificio Euro n° 24 - 5°F,
Infantado
2670-391 Loures
11015452
N1676CA60L01/0714
- Olho Seco

Olho Seco
THEAHYABAK

THEAHYABAK®
Este produto é um dispositivo médico. Por favor leia as
instruções antes de utilizar.
Conserve este folheto. Pode ter necessidade de o ler novamente.
INDICAÇÕES
THEAHYABAK destina-se a hidratar e lubrificar o olho em caso de sensação de secura ou olhos cansados.
THEAHYABAK destina-se também a lubrificar as lentes de contacto em caso de desconforto.
Apenas para uso oftálmico.
ADEQUADO PARA
- Adultos,
- Crianças: o produto deve ser administrado pelos pais ou cuidadores,
- Mulheres grávidas ou a amamentar, com base no conhecimento atual,
- Utilizadores de qualquer tipo de lentes de contacto.
CONTRAINDICAÇÕES
Não utilize THEAHYABAK se é alérgico a qualquer um dos seus ingredientes (ver descrição do dispositivo).
DESCRIÇÃO DO DISPOSITIVO
THEAHYABAK é uma solução oftálmica composta por ácido hialurónico (sob a forma de hialuronato de sódio 0,15%), cloreto de sódio, trometamol, ácido clorídrico e água para preparações injetáveis.
THEAHYABAK é uma solução estéril, isenta de fosfatos, isotónica e de pH neutro.
A solução é fornecida num frasco multidose ABAK®. Este dispositivo inovador e patenteado fornece a solução oftálmica através de um filtro de 0,2 µm, prevenindo assim qualquer contaminação bacteriana da solução, sem a utilização de quaisquer conservantes.
Desta forma, THEAHYABAK é suave para os olhos uma vez que não contém conservantes. A ausência de conservantes permite proteger os tecidos oculares e não provoca reações em utilizadores sensíveis aos conservantes.
BENEFÍCIOS, DESEMPENHO E MECANISMO DE AÇÃO
THEAHYABAK é recomendado para a hidratação e lubrificação do olho em caso de sensação de secura ou fadiga ocular provocadas por fatores externos tais como vento, fumo, poluição, poeiras, calor seco, ar condicionado, viagem de avião ou trabalho prolongado à frente de um monitor de computador. Proporciona, assim, um alívio imediato e um maior conforto para os olhos durante o dia.
THEAHYABAK é também recomendado para utilizadores de lentes de contacto. Lubrifica e hidrata as lentes, facilitando a sua colocação ou remoção, e permitindo a sua utilização ao longo do dia.
O principal componente do THEAHYABAK é o ácido hialurónico (sob a forma de hialuronato de sódio), um polissacarídeo natural presente no olho humano que retém a água, assegurando a hidratação e lubrificação da superfície do olho. O hialuronato de sódio mantém a solução na superfície do olho proporcionando um alívio duradouro e reduzindo o tempo de cicatrização da superfície ocular.
RECOMENDAÇÕES GERAIS DE UTILIZAÇÃO
Não é necessário qualquer treino específico para utilizar THEAHYABAK.
Lave cuidadosamente as mãos antes de utilizar.
Não utilize qualquer acessório para abrir o frasco.
Quando se inicia uma embalagem de THEAHYABAK, a primeira gota demora mais a surgir devido ao sistema de filtro específico do frasco (ver descrição do dispositivo).
Volte a colocar a tampa no frasco após a utilização.
Para hidratação e lubrificação do olho:
Posologia: 1 gota em cada olho, sempre que necessário ao longo do dia.
Aplique 1 gota dentro da bolsa formada pela pálpebra inferior, puxando ligeiramente a pálpebra inferior para baixo e olhando para cima. Feche suavemente o olho após a aplicação.

Para lubrificação e hidratação das lentes de contacto:
Posologia: 1 gota em cada lente de contacto ao colocar ou retirar as mesmas, tão frequentemente quanto necessário ao longo do dia.
Antes de colocar as suas lentes de contacto, aplique uma gota na face interna da lente quando esta se encontra sobre o seu dedo.
Coloque as suas lentes como habitualmente e pestaneje algumas vezes para permitir que a solução estabilize, assegurando assim uma distribuição uniforme de THEAHYABAK sobre o olho e a superfície das lentes de contacto.

PRECAUÇÕES DE UTILIZAÇÃO
Produto de utilização múltipla para um único doente.
Não partilhe frascos com outras pessoas a fim de evitar contaminação cruzada.
Verifique se a embalagem está intacta antes da primeira utilização deste produto.
Assegure-se de que o anel de segurança da tampa do frasco está intacto antes de o utilizar pela primeira vez.
Para evitar qualquer contaminação, não toque com a ponta do frasco em nenhuma superfície.
Deve contactar um profissional de saúde se os seus sintomas se agravarem ou se não melhorar.
ADVERTÊNCIAS
Mantenha fora da vista e do alcance das crianças.
Não injete nem ingira.
Após a utilização, não conduza ou utilize máquinas se a sua visão estiver turva: aguarde alguns minutos até que a sua visão recupere.
Aguarde pelo menos 10 minutos antes de utilizar produtos oftálmicos diferentes.
Nunca dilua o THEAHYABAK nem o misture com outras soluções.
Se sujeito a baixa pressão atmosférica, o frasco pode verter o seu conteúdo.
EFEITOS SECUNDÁRIOS
Foram notificados os seguintes efeitos secundários: possibilidade rara de irritação ocular ligeira, reações alérgicas, dor no olho, sensação de corpo estranho, visão turva e vermelhidão ocular. Nestas situações, os utilizadores de lentes de contacto devem retirar as suas lentes de contacto.
Quando utiliza soluções oftálmicas, pode ter sintomas que provocam incómodo tais como sensação de ardor no olho ou sensação de picada no olho durante um curto período de tempo.
Se detetar quaisquer efeitos secundários ou qualquer sensação estranha após a utilização deste produto, por favor contacte um profissional de saúde e notifique o distribuidor local, o fabricante ou as Autoridades de Saúde locais (ver os dados de contacto no final das instruções de utilização).
CONDIÇÕES DE CONSERVAÇÃO E ELIMINAÇÃO
Conservar entre 8 ºC e 30 ºC.
Não exponha o produto à luz solar direta ou a humidade.
Não utilize após o prazo de validade, impresso no frasco e na embalagem exterior.
O prazo de validade corresponde ao último dia do mês indicado, desde que a embalagem permaneça intacta e tenha sido adequadamente conservada.
O produto pode ser utilizado até 6 meses após a primeira abertura. Após este prazo, elimine o produto remanescente de forma responsável.
Elimine o produto de forma responsável e de acordo com as orientações locais (o frasco deve ser tratado como lixo doméstico e a cartonagem e o folheto são recicláveis).
Data de preparação e número de versão das instruções de utilização: Ver.5 11/2023
THEAHYABAK® é uma marca registada dos Laboratoires Théa.
©2024, Laboratoires Théa. All rights reserved.
Fabricante:
Laboratoires Théa
12 rue Louis Blériot
63017 Clermont-Ferrand
Cedex 2 France
www.laboratoires-thea.com
Seção « Contact us

Distribuidor:
Théa Portugal, S.A.
Rua Pedro Álvares Cabral,
nº 24 - 5º F
2670-391 Loures - Portugal
Autoridade de Saúde Competente:
INFARMED - Autoridade Nacional do Medicamento e Produtos de Saúde,
I.P. Parque de Saúde de Lisboa, Avenida do Brasil 53, 1749-004 Lisboa
Tel: +351 217 987 100 – Fax: +351 217 987 316
e-mail: infarmed@infarmed.pt – www.infarmed.pt

SIGNIFICADO DOS SÍMBOLOS DA EMBALAGEM


Olho Seco
Siccafluid

FOLHETO INFORMATIVO: INFORMAÇÃO PARA O UTILIZADOR
SICCAFLUID 2,5 mg/g gel oftálmico
Carbómero 974P
Leia com atenção todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico ou farmacêutico.
-Conserve este folheto. Pode ter necessidade de o ler novamente.
-Caso precise de esclarecimentos ou conselhos, consulte o seu farmacêutico.
-Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico. Ver secção 4.
-Se não se sentir melhor ou se piorar, tem de consultar um médico.
Neste folheto:
1. O que é SICCAFLUID e para que é utilizado?
2. Antes de utilizar SICCAFLUID
3. Como utilizar SICCAFLUID
4. Efeitos secundários possíveis
5. Como conservar SICCAFLUID
6. Outras informações
1. O QUE É SICCAFLUID E PARA QUE É UTILIZADO
SICCAFLUID é um substituto das lágrimas que contém um lubrificante denominado Carbómero 974P.
Trata-se de um gel oftálmico utilizado no alívio dos sintomas de olho seco (como dor, ardor, irritação ou secura) causados pelo facto dos seus olhos não produzirem lágrimas suficientes.
2. Antes de utilizar SICCAFLUID
Não utilize SICCAFLUID
- se tem alergia (hipersensibilidade) ao Carbómero ou a qualquer outro componente de SICCAFLUID indicado na Secção 6, “Qual a composição de SICCAFLUID”.
Advertências e precauções
- se utiliza lentes de contacto moles: tem de retirá-las antes de utilizar SICCAFLUID gel oftálmico. Após utilizar SICCAFLUID, espere pelo menos 30 minutos antes de voltar a colocar as lentes de contacto. Ver também Secção 2, “SICCAFLUID contém cloreto de benzalcónio”.
- se sentir um agravamento ou uma persistência dos sintomas após iniciar o tratamento com SICCAFLUID: Contacte o seu médico.
- NÃO INJETE, NÃO INGIRA.
Crianças e adolescentes até aos 18 anos de idade:
A segurança e a eficácia do SICCAFLUID em crianças e adolescentes na posologia recomendada para adultos, foram estabelecidas através da experiência clínica, mas não estão disponíveis ensaios clínicos.
Outros medicamentos e SICCAFLUID
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos obtidos sem receita médica.Se tem de utilizar qualquer outro medicamento a nível ocular durante o tratamento com SICCAFLUID: primeiro utilize o outro medicamento, espere 15 minutos, e depois utilize SICCAFLUID.
Gravidez e amamentação
Se está grávida ou a amamentar, consulte o seu médico antes de utilizar SICCAFLUID.
O seu médico irá decidir se pode utilizar SICCAFLUID.
Condução de veículos e utilização de máquinas
A sua visão pode tornar-se turva durante um curto espaço de tempo após utilizar SICCAFLUID. Não deve conduzir ou utilizar máquinas até que a sua visão volte ao normal.
SICCAFLUID contém cloreto de benzalcónio
Este medicamento contém 0,0015 mg de cloreto de benzalcónio em cada gota que é equivalente a 0,06 mg/g.
O cloreto de benzalcónio pode ser absorvido pelas lentes de contacto moles e pode alterar a cor das lentes de contacto. Deve remover as lentes de contacto antes da utilização deste medicamento e voltar a colocá-las 30 minutos depois.
O cloreto de benzalcónio pode também causar irritação nos olhos, especialmente se tem os olhos secos ou alterações da córnea (a camada transparente na parte da frente do olho). Se tiver uma sensação estranha no olho, picadas ou dor no olho apos utilizar este medicamento, fale com o seu médico.
3. Como utilizar SICCAFLUID
Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico ou farmacêutico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
A dose habitual de SICCAFLUID é de 1 gota em cada olho que necessita de tratamento, até 4 vezes por dia.
Crianças e adolescentes até aos 18 anos de idade:
A segurança e a eficácia do SICCAFLUID em crianças e adolescentes na posologia recomendada para adultos, foram estabelecidas através da experiência clínica, mas não estão disponíveis ensaios clínicos.
Modo de administração
Não utilize o frasco se o selo protetor da tampa estiver danificado antes da primeira utilização.
Lave as mãos antes de abrir o frasco. Incline a sua cabeça para trás e olhe para cima.
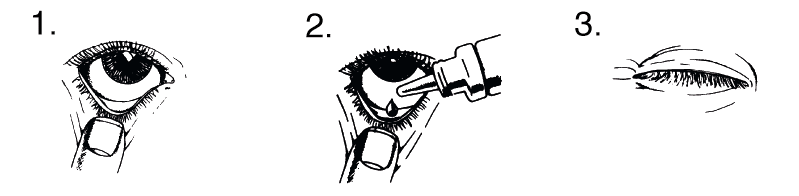
1. Puxe cuidadosamente para baixo a pálpebra inferior do olho que necessita de tratamento até que se forme uma pequena “bolsa”.
2. Inverta o frasco. Aperte-o para colocar 1 gota na “bolsa”.
3. Solte a pálpebra inferior e pestaneje algumas vezes.
4. Repita os passos 1 a 3 para o outro olho, se este também necessitar de tratamento.
Para prevenir uma infeção evite tocar com a extremidade do frasco no olho, no tecido envolvente ou em outro local qualquer. Coloque novamente a tampa de rosca para fechar o frasco, imediatamente após a sua utilização.
Conserve o frasco na vertical com a tampa virada para baixo, de modo a facilitar a formação das gotas na utilização seguinte de SICCAFLUID.
Uma vez aberto o frasco, não deve utilizá-lo mais de 28 dias; ver também Secção 5, “Como conservar SICCAFLUID”.
Se utilizar mais SICCAFLUID do que deveria
A aplicação de mais gotas de SICCAFLUID do que deveria não representa qualquer perigo.
Caso se tenha esquecido de utilizar SICCAFLUID
Caso se tenha esquecido de utilizar uma dose de SICCAFLUID continue com a dose seguinte como habitualmente.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com seu médico ou farmacêutico.
4. Efeitos Secundários Possívies
Como todos os medicamentos, SICCAFLUID pode causar efeitos secundários, no entanto estes não se manifestam em todas as pessoas.
Contacte o seu médico se:
- sentir um agravamento ou uma persistência dos sintomas após iniciar o tratamento com SICCAFLUID.
Se sentir algum dos seguintes efeitos secundários logo após aplicar o gel oftálmico, fale com o seu médico caso os sintomas o incomodem:
- visão turva temporária
- sensação de ardor ou de queimadura ligeira e transitória no olho.
Sabe-se que ocorrem os sintomas acima mencionados, mas o número de pessoas que são afetadas pode variar.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet:
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. Como conservar SICCAFLUID
Manter fora do alcance e da vista das crianças.
Não utilize SICCAFLUID após o prazo de validade impresso no rótulo do frasco e na embalagem exterior após EXP. O prazo de validade corresponde ao último dia do mês indicado.
Não conservar acima de 25ºC.
Manter o frasco dentro da embalagem exterior, para proteger da luz.
Conserve o frasco na vertical com a tampa virada para baixo, de modo a facilitar a formação das gotas na utilização seguinte de SICCAFLUID.
Deve rejeitar o frasco 28 dias após a primeira abertura, mesmo que ainda contenha algumas gotas.
Os medicamentos não devem ser eliminados na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como eliminar os medicamentos de que já não necessita. Estas medidas irão ajudar a proteger o ambiente.
6. Conteúdo da embalagem e outras informações
Qual a composição de SICCAFLUID
- A substância ativa é o Carbómero 974P 2,5 mg/g.
- Os outros componentes são o cloreto de benzalcónio, sorbitol, lisina mono-hidratada, acetato de sódio tri-hidratado, álcool polivinílico, água para preparações injetáveis.
Qual o aspeto de SICCAFLUID e conteúdo da embalagem
SICCALFUID é um gel ligeiramente amarelado e opalescente que se apresenta num frasco de plástico com uma tampa de rosca.
Cada frasco contém 10 g de gel oftálmico.
Cada embalagem contém 1 frasco.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado
Laboratoires THEA
12, rue Louis Blériot
63017 CLERMONT- FERRAND Cedex 2
FRANÇA
Fabricante
URSAPHARM, Arzneimittel GmbH & Co. KG
Industriestrasse
66129 Saarbrücken
ALEMANHA
Este folheto foi aprovado pela última vez em 03/2021
FOLHETO INFORMATIVO: INFORMAÇÃO PARA O UTILIZADOR
SICCAFLUID 2,5 mg/g gel oftálmico em recipiente unidose
Carbómero 974P
Leia com atenção todo este folheto antes de começar a utilizar este medicamento pois contém informação importante para si.
Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico ou farmacêutico.
-Conserve este folheto. Pode ter necessidade de o ler novamente.
-Caso precise de esclarecimentos ou conselhos, consulte o seu farmacêutico.
-Se tiver quaisquer efeitos secundários, incluindo possíveis efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico. Ver secção 4.
-Se não se sentir melhor ou se piorar, tem de consultar um médico.
Neste folheto:
1. O que é SICCAFLUID e para que é utilizado
2. Antes de utilizar SICCAFLUID
3. Como utilizar SICCAFLUID
4. Efeitos secundários possíveis
5. Como conservar SICCAFLUID
6. Outras informações
1. O QUE É SICCAFLUID E PARA QUE É UTILIZADO.
SICCAFLUID é um substituto das lágrimas que contém um lubrificante denominado Carbómero 974P.
Trata-se de um gel oftálmico utilizado no alívio dos sintomas de olho seco (como dor, ardor, irritação ou secura) causados pelo facto dos seus olhos não produzirem lágrimas suficientes.
2. ANTES DE UTILIZAR SICCAFLUID
Não utilize SICCAFLUID
- se tem alergia (hipersensibilidade) ao Carbómero ou a qualquer outro componente de SICCAFLUID indicado na Secção 6, “Qual a composição de SICCAFLUID”.
Tome especial cuidado com SICCAFLUID
- se sentir um agravamento ou uma persistência dos sintomas após iniciar o tratamento com SICCAFLUID: Contacte o seu médico.
- NÃO INJETE, NÃO INGIRA.
Ao utilizar SICCAFLUID com outros medicamentos
Informe o seu médico ou farmacêutico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos obtidos sem receita médica.
Se tem de utilizar qualquer outro medicamento a nível ocular durante o tratamento com SICCAFLUID: primeiro utilize o outro medicamento, espere 15 minutos, e depois utilize SICCAFLUID.
Gravidez e aleitamento
Se está grávida ou a amamentar consulte o seu médico antes de utilizar SICCAFLUID.
O seu médico irá decidir se pode utilizar SICCAFLUID.
Crianças
Não se recomenda a utilização de SICCAFLUID em crianças. Para mais informações consulte o seu médico ou farmacêutico.
Condução de veículos e utilização de máquinas
A sua visão pode tornar-se turva durante um curto espaço de tempo após utilizar SICCAFLUID. Não deve conduzir ou utilizar máquinas até que a sua visão volte ao normal.
3. COMO UTILIZAR SICCAFLUID
Utilize este medicamento exatamente como está descrito neste folheto, ou de acordo com as indicações do seu médico ou farmacêutico. Fale com o seu médico ou farmacêutico se tiver dúvidas.
A dose habitual de SICCAFLUID é de 1 gota em cada olho que necessita de tratamento, até 4 vezes por dia.
Modo de administração:
Lave as mãos antes de abrir o recipiente unidose.
Certifique-se que o gel está na ponta do recipiente. Para abrir o recipiente rode a aba.
Incline a sua cabeça para trás e olhe para cima.
1. Puxe cuidadosamente para baixo a pálpebra inferior do olho que necessita de tratamento até que se forme uma pequena “bolsa”.
2. Vire o recipiente unidose para baixo. Aperte-o para colocar 1 gota na “bolsa”.
3. Solte a pálpebra inferior e pestaneje algumas vezes.
4. Repita os passos 1 a 3 para o outro olho, se este também necessitar de tratamento. Um recipiente unidose é suficiente para tratar os dois olhos, caso seja necessário.
Para prevenir uma infeção evite o contacto da ponta do recipiente unidose com o olho, o tecido envolvente ou outro local qualquer.
Rejeite o recipiente unidose após a utilização. Não conserve o recipiente para uma utilização posterior.
Se utilizar mais SICCAFLUID do que deveria
A aplicação de mais gotas de SICCAFLUID do que deveria não representa qualquer perigo.
Caso se tenha esquecido de utilizar SICCAFLUID
Caso se tenha esquecido de utilizar uma dose de SICCAFLUID continue com a dose seguinte como habitualmente.
Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com seu médico ou farmacêutico.
4. EFEITOS SECUNDÁRIOS POSSÍVEIS
Como todos os medicamentos, SICCAFLUID pode causar efeitos secundários, no entanto estes não se manifestam em todas as pessoas.
Contacte o seu médico se:
- sentir um agravamento ou uma persistência dos sintomas após iniciar o tratamento com SICCAFLUID.
Se sentir algum dos seguintes efeitos secundários logo após aplicar o gel oftálmico, fale com o seu médico caso os sintomas o incomodem:
- visão turva temporária
- sensação de ardor ou de queimadura ligeira e transitória no olho.
Sabe-se que ocorrem os sintomas acima mencionados, mas o número de pessoas que são afetadas pode variar.
Comunicação de efeitos secundários
Se tiver quaisquer efeitos secundários, incluindo efeitos secundários não indicados neste folheto, fale com o seu médico ou farmacêutico. Também poderá comunicar efeitos secundários diretamente ao INFARMED, I.P. através dos contactos abaixo. Ao comunicar efeitos secundários, estará a ajudar a fornecer mais informações sobre a segurança deste medicamento.
INFARMED, I.P.
Direção de Gestão de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
Sítio da internet:
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
E-mail: farmacovigilancia@infarmed.pt
5. COMO CONSERVAR SICCAFLUID
Manter fora do alcance e da vista das crianças.
Não utilize SICCAFLUID após o prazo de validade impresso no recipiente unidose, na saqueta e na embalagem exterior após “EXP”. O prazo de validade corresponde ao último dia do mês indicado.
Não conservar acima de 25ºC.
Manter os recipientes unidose dentro da embalagem exterior, para proteger da luz.
Após utilizar, rejeite imediatamente o recipiente unidose que tenha sido aberto com a solução que ainda reste. Não conserve para uma futura utilização.
Os medicamentos não devem ser eliminados na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como eliminar os medicamentos de que já não necessita. Estas medidas irão ajudar a proteger o ambiente.
6. OUTRAS INFORMAÇÕES
Qual a composição de SICCAFLUID
- A substância ativa é o carbómero 974P 2,5 mg/g.
- Os outros componentes são: sorbitol, lisina mono-hidratada, acetato de sódio tri-hidratado, álcool polivinílico e água para preparações injetáveis.
Qual o aspeto de SICCAFLUID e conteúdo da embalagem
SICCAFLUID é um gel ligeiramente amarelado e opalescente em recipiente unidose.
Cada recipiente unidose contém 0,5 g de gel oftálmico. A embalagem contém 10, 20, 30 ou 60 recipientes unidose.
É possível que não sejam comercializadas todas as apresentações.
Titular da Autorização de Introdução no Mercado e Fabricante
Titular da Autorização de Introdução no Mercado:
LABORATOIRES THEA
12, RUE LOUIS BLERIOT
63017 CLERMONT-FERRAND CEDEX 2
FRANÇA
Fabricante:
LABORATOIRE UNITHER
ESPACE INDUSTRIEL NORD
151 RUE ANDRE DUROUCHEZ
80084 AMIENS CEDEX 2 – BP 28028
FRANÇA
Ou
LABORATOIRE UNITHER
1 rue de l’Arquerie
50200COUTANCES
FRANÇA
Este medicamento encontra-se autorizado nos Estados Membros da UE com os seguintes nomes:
França, Espanha, Itália e Portugal: SICCAFLUID
Holanda: SICCAFLUID UNIDOSE
Islândia e Noruega: OFTAGEL
Este folheto foi aprovado pela última vez em: 10/2021

Olho Seco
Thealoz Duo

Thealoz Duo
Proteção, hidratação e lubrificação dos olhos para o tratamento do olho seco moderado a grave
Frasco ABAK® - Sem conservantes.
COMPOSIÇÃO:
Trealose, 3g Hialuronato de sódio, 0,15g Cloreto de sódio, trometamol, ácido clorídrico, água para preparações injetáveis para 100ml.
NOME E MORADA DO FABRICANTE:
Laboratoires THEA
12 rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França
INDICAÇÕES:
THEALOZ DUO contém uma solução que pode ser aplicada nos olhos ou nas lentes de contacto.
THEALOZ DUO é recomendado em casos de desconforto, sensação de picada e irritação dos olhos.
Estes sintomas podem ser causados, por exemplo, por fatores externos tais como vento, fumo, poluição, poeira, exposição ao sol ou ao frio, calor seco, ar condicionado, viagens de avião, várias horas a trabalhar em frente ao computador…
Uma vez que não contém conservantes, THEALOZ DUO mantém a integridade dos tecidos oculares, pelo que também pode ser utilizado pelos portadores de lentes de contacto de qualquer tipo.
Proporciona um conforto imediato que se mantém ao longo de todo o dia.
PROPRIEDADES:
THEALOZ DUO é uma solução oftálmica aquosa estéril, sem conservantes, hipotonica e com pH neutro.
Os seus principais componentes são trealose, um dissacárido presente em muitas plantas e animais, e hialuronato de sódio, um polissacárido presente naturalmente no olho.
As características físico-químicas da trealose conferem propriedades protetoras, antioxidantes e hidratantes.
Constitui assim um elemento essencial no mecanismo anidrobiótico observado em certos microrganismos (um estado metabólico mais lento que lhes permite sobreviver em situações de desidratação extrema).
A trealose também possui propriedades que permitem a proteção e a estabilização das membranas celulares, evitando a alteração de proteínas e a deterioração de lípidos, bem como propriedades antioxidantes.
O hialuronato de sódio é um polissacárido que existe no olho humano e que possui propriedades únicas de retenção de água.
As suas importantes propriedades de retenção de água resultam num aumento da hidratação e lubrificação da superfície da córnea.
Para além disso, as suas propriedades bioadesivas conferem uma melhor aderência do produto à superfície do olho, o que permite um alívio de longa duração e uma diminuição do tempo necessário para cicatrização da córnea.
Deste modo, graças à associação única de trealose e hialuronato de sódio, o THEALOZ DUO assegura a proteção, hidratação e lubrificação da superfície ocular por um maior período de tempo e com um máximo conforto.
Para uma tolerância máxima, THEALOZ DUO é uma solução que não contém conservantes, graças ao frasco ABAK® multidose.
Este dispositivo inovador e registado sob patente permite a instilação de gotas oftálmicas através de uma membrana com um filtro de 0,2 µm, o que previne a contaminação da solução por bactérias.
A solução fica assim protegida e pode ser utilizada até 6 meses após abertura do frasco.
- ADVERTÊNCIAS E PRECAUÇÕES ESPECIAIS DE UTILIZAÇÃO:
- Não utilizar se for alérgico a qualquer um dos componentes.
- Não tocar na superfície dos olhos com a extremidade do frasco.
- Não injetar, não engolir.
- Não utilizar o produto caso o selo de segurança esteja danificado.
MANTER FORA DA VISTA E DO ALCANCE DAS CRIANÇAS.
INTERAÇÕES:
Deve haver um intervalo de, pelo menos, 10 minutos entre a aplicação de dois produtos oftálmicos diferentes.
COMO UTILIZAR ESTE PRODUTO:
MODO E VIA DE ADMINISTRAÇÃO: INSTILAÇÃO OCULAR.
- Lavar cuidadosamente as mãos antes de usar.
- Evitar tocar com a extremidade do frasco nos olhos ou nas pálpebras, especialmente se tiver alguma infeção ocular.
- Instilar uma gota no saco conjuntival inferior, olhando para cima e puxando ligeiramente a pálpebra para baixo.
- Voltar a colocar a tampa após a utilização. POSOLOGIA: 1 gota em cada olho, 4 a 6 vezes por dia. THEALOZ DUO pode ser utilizado com as lentes de contacto colocadas.
EFEITOS SECUNDÁRIOS INDESEJÁVEIS E DESAGRADÁVEIS (EFEITOS ADVERSOS):
Rara possibilidade de irritação ocular ligeira.
CONSERVAÇÃO DO PRODUTO:
Não conservar o frasco aberto por mais de 6 meses. Conservar a temperatura inferior a 25°C.
NÃO UTILIZAR APÓS O PRAZO DE VALIDADE INDICADO NA EMBALAGEM.
O prazo de validade refere-se ao produto na embalagem original e conservado de forma correta.
Não utilizar caso o frasco esteja danificado.
REVISÃO DO TEXTO: 02/2013

Olho Seco
Thealoz Duo Gel

Thealoz Duo Gel
Proteção, hidratação e lubrificação do olho para o alívio dos sintomas de olho seco.
Tratamento da síndrome do olho seco moderado a grave.
Recipientes unidose - Sem conservantes.
COMPOSIÇÃO:
Trealose 3g, Hialuronato de sódio 0,15g, Carbómero (Carbopol 974P) 0,25g, Sorbitol, hidróxido de sódio, água para injetáveis qbp 100g.
NOME E MORADA DO FABRICANTE:
Laboratoires Théa
12 rue Louis Blériot 63017
Clermont-Ferrand - Cedex 2
França
EM QUE CASOS SE DEVE UTILIZAR THEALOZ DUO GEL:
THEALOZ DUO GEL é um gel fluido para ser aplicado na superfície do olho durante o dia e durante a noite.
Pode utilizar THEALOZ DUO GEL quando tiver sinais ou sintomas moderados a graves de olho seco tais como desconforto, sensação de picadas ou irritação no olho.
Estes sintomas podem ser causados por fatores externos como vento, fumo, poluição, pó, sol ou situações de frio, calor seco, ar condicionado, viagens de avião ou trabalho prolongado frente a um ecrã de computador.
THEALOZ DUO GEL não tem conservantes e previne a irritação e as reações alérgicas que podem ocorrer quando são utilizados produtos para aplicação ocular com conservantes.
É recomendada a utilização de produtos sem conservantes quando se tem olhos sensíveis ou quando se utiliza os produtos durante um longo período de tempo.
PROPRIEDADES:
THEALOZ DUO GEL é um gel fluido, estéril, sem conservantes, hipotónico e de pH neutro, o que significa que tem o mesmo pH que a superfície do olho e ajuda a manter a hidratação na sua superfície.
Os seus principais componentes são a trealose, uma molécula presente em várias plantas e animais, o hialuronato de sódio, outra molécula presente de forma natural no olho e o carbómero (Carbopol 974P) que permite que o THEALOZ DUO GEL permaneça mais tempo no olho.
A trealose protege, hidrata e tem propriedades antioxidantes.
Por estes motivos, constitui um elemento essencial no mecanismo de anidrobiose utilizado por certos organismos de pequenas dimensões (um estado metabólico lento que lhes permite sobreviver em condições de desidratação).
A trealose também protege as células através da estabilização das suas membranas.
O hialuronato de sódio é um polissacárido de origem natural (molécula complexa) que é encontrado no olho humano e que ajuda a manter a água, hidratando e lubrificando a superfície do olho.
Mantém o gel na superfície do olho, proporcionando um alívio durador e diminuindo o tempo de cicatrização do epitélio da córnea (camada transparente do olho).
O carbómero permite que o THEALOZ DUO GEL permaneça na superfície do olho durante mais tempo, proporcionando um alívio durador dos sinais e sintomas de olho seco.
A combinação especial de trealose, hialuronato de sódio e carbómero no THEALOZ DUO GEL hidrata e lubrifica, mas também protege a superfície do olho durante mais tempo e com maior conforto.
ADVERTÊNCIAS E PRECAUÇÕES ESPECIAIS DE UTILIZAÇÃO:
- Não utilize se é alérgico a qualquer dos componentes.
- Não toque na superfície do olho com a ponta do recipiente unidose.
- Não injete, não ingira.
- Não utilize se o recipiente estiver danificado.
MANTER FORA DA VISTA E DO ALCANCE DAS CRIANÇAS.
INTERAÇÕES:
Espere pelo menos 15 minutos entre a utilização de dois colírios diferentes.
Utilize o THEALOZ DUO GEL em último lugar.
COMO UTILIZAR THEALOZ DUO GEL:
MODO E VIA DE ADMINISTRAÇÃO:
UTILIZE APENAS NO OLHO.
- Lave bem as mãos antes de utilizar.
- Retire um recipiente unidose da tira (figura 1).
- Vire o recipiente unidose para baixo e bata suavemente no recipiente.
- Abra o recipiente e pressione para libertar o gel (figura 2).
- Instile uma gota no saco conjuntival (o espaço entre o olho e a pálpebra), enquanto puxa suavemente a pálpebra inferior para baixo e olha para cima (figura 3).
- Rejeite o recipiente, mesmo que ainda reste algum gel.
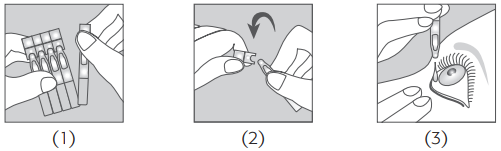
Caso utilize lentes de contacto, deve retirá-las antes de utilizar o THEALOZ DUO GEL.
Depois de utilizar o THEALOZ DUO GEL, aguarde pelo menos 30 minutos antes de voltar a colocar as lentes.
POSOLOGIA:
1 gota em cada olho, 2 a 4 vezes ao dia.
EFEITOS SECUNDÁRIOS POSSÍVEIS:
Apesar de ser pouco frequente, pode sentir uma ligeira irritação no olho.
COMO CONSERVAR THEALOZ DUO GEL:
Conserve os recipientes unidose na embalagem exterior para os proteger da exposição direta à luz.
Conservar a temperatura inferior a 25ºC.
NÃO UTILIZAR APÓS O PRAZO DE VALIDADE INDICADO NA EMBALAGEM EXTERIOR.
O prazo de validade corresponde ao produto acondicionado na embalagem intacta e corretamente conservada.
Não utilize se os recipientes estiverem danificados.
Rejeite os recipientes abertos imediatamente após a utilização.
Não os deite fora no lixo doméstico.
ÚLTIMA REVISÃO DAS INSTRUÇÕES DE UTILIZAÇÃO: 03/2014.
- Pálpebras

Pálpebras
Blephasteam

INSTRUÇÕES DE UTILIZAÇÃO
BLEPHASTEAM®
Dispositivo para aquecimento do olho, tecnologia de calor húmido
ÍNDICE
I. DESCRIÇÃO DO DISPOSITIVO
II. INDICAÇÕES
III. ADVERTÊNCIAS
IV. CONTRA-INDICAÇÕES
V. PRECAUÇÕES DE UTILIZAÇÃO E ADVERTÊNCIAS
VI. RECOMENDAÇÕES PARA A SUA UTILIZAÇÃO
VII. INSTRUÇÕES PARA UTILIZAR O DISPOSITIVO BLEPHASTEAM®
VIII. LIMPAR E GUARDAR O SEU DISPOSITIVO BLEPHASTEAM®
IX. GARANTIA E SERVIÇOS
X. MANUTENÇÃO E INSTRUÇÕES EM CASO DE AVARIA
XI. FABRICANTE E DISTRIBUIDORES NA EUROPA
XII. INFORMAÇÕES TÉCNICAS
I. DESCRIÇÃO DO DISPOSITIVO BLEPHASTEAM®
é um dispositivo médico patenteado, baseado nos resultados da pesquisa científica do Dr J R Fuller.
Este dispositivo inovador proporciona um ambiente que aumenta naturalmente a qualidade da lágrima e a sua estabilidade, reforçando a espessura da camada lipídica do filme lacrimal.
Esta camada evita a evaporação do filme lacrimal; é produzida pelas glândulas de Meibomius que se encontram nas pálpebras.
Estas glândulas podem obstruir-se com facilidade, criando patologias como a síndrome do olho seco.
BLEPHASTEAM® proporciona uma terapia de calor latente que torna as secreções menos espessas e desbloqueia as glândulas de Meibomius, melhorando a estabilidade do filme lacrimal.
Devido a este efeito, BLEPHASTEAM® melhora a saúde das pálpebras e o conforto diário.
BLEPHASTEAM® foi testado e validado de forma a tornar segura a sua utilização.
Durante a utilização de BLEPHASTEAM®, a visão mantém-se nítida e a função das pálpebras permanece normal; tal promove a secreção do filme lacrimal lipídico e o aumento da produção de lágrimas quando pestaneja.
A temperatura atingida a nível da córnea é de 42,5°C; esta temperatura mantém-se constante durante a utilização.
Os componentes do dispositivo estão permanentemente ligados.

BLEPHASTEAM®
Importante:
BLEPHASTEAM® deve permanecer ligado durante 15 minutos antes da sua utilização para permitir um correto pré-aquecimento (ver instruções de utilização na secção VII).
As luzes amarela e verde na caixa de controlo indicam o seguinte:
Amarela
O dispositivo está em pré-aquecimento.
Verde contínua
O pré-aquecimento terminou e o dispositivo está pronto para uma sessão de tratamento.
Verde intermitente
O dispositivo está a ser utilizado (tratamento em curso) e o cronómetro está ligado há 10 minutos.
II. INDICAÇÕES BLEPHASTEAM®
BLEPHASTEAM® alivia os sintomas associados à disfunção das glândulas de Meibomius.
Esta disfunção provoca diversas patologias como a síndrome do olho seco.
O calor e a humidade proporcionados pelo dispositivo diminuem a espessura das secreções que provocam a obstrução destas glândulas tornando mais fácil remover as secreções através de uma massagem ou exercendo pressão.
Para quaisquer questões acerca deste dispositivo ou para aconselhamento médico, por favor contacte o seu oftalmologista ou a THÉA PORTUGAL (ver contactos no final deste folheto).
III. ADVERTÊNCIAS BLEPHASTEAM®
só pode ser utilizado por crianças sob a supervisão de um adulto e após consultar o seu oftalmologista.
Se utiliza lentes de contacto ou óculos, retire-os sempre antes de utilizar BLEPHASTEAM®.
Se tiver uma reação adversa ao BLEPHASTEAM®, como uma sensibilidade excessiva ao calor ou uma reação alérgica a um dos componentes do dispositivo, pare imediatamente o tratamento e consulte o seu médico.
Não deverá administrar nenhum colírio durante, pelo menos, 15 minutos antes de utilizar BLEPHASTEAM®.
IV. CONTRA-INDICAÇÕES BLEPHASTEAM®
não deve ser utilizado nos seguintes casos, exceto se indicado pelo seu médico:
- Doenças agudas dos olhos e/ou das pálpebras,
- Ferimentos recentes e lesões na córnea ou no olho,
- Seborreia Meibomiana (secreção sebácea excessiva nas glândulas de Meibomius),
- Após cirurgia do olho ou em redor do olho.
V. PRECAUÇÕES DE UTILIZAÇÃO E ADVERTÊNCIAS
Leia cuidadosamente antes de utilizar
Deve seguir estas instruções quando utilizar este dispositivo médico.
Caso contrário, poderá colocar em risco a segurança e o funcionamento do equipamento e danificar o dispositivo.
PRECAUÇÕES DE UTILIZAÇÃO
◦ Este dispositivo não deve ser utilizado por nenhum doente (incluindo crianças) que tenha as suas capacidades físicas, sensoriais ou mentais diminuídas, exceto sob supervisão e com aconselhamento de um oftalmologista.
◦ Este dispositivo médico só deve ser utilizado por um adulto capaz ou sob a supervisão de um adulto capaz quando for utilizado por uma criança ou por determinados adultos que requerem supervisão.
◦ Manter o dispositivo fora do alcance das crianças.
◦ Quando utilizado por crianças, BLEPHASTEAM® deve ser usado na presença de um adulto.
ADVERTÊNCIAS
- Não coloque ou utilize este dispositivo em locais onde poderá entrar em contacto com líquidos ou ser exposto a condições ambientais, tais como temperaturas muito elevadas, humidade, exposição direta à luz solar, pó, sal, etc., que poderão danificá-lo.
- Não instale ou utilize este dispositivo na presença de gás ou produtos químicos inflamáveis ou explosivos.
- Durante a sua utilização, não bloqueie o acesso à tomada elétrica no caso de ser necessário retirar rapidamente a ficha da tomada.
- Retire a ficha elétrica da tomada puxando pela ficha e não pelo cabo pois poderá danificar a ficha.
- Não exponha o dispositivo ou os anéis a temperaturas muito elevadas nem coloque perto de uma chama.
- Não coloque o dispositivo dentro de água nem lave com água corrente.
- Não limpe com quaisquer produtos químicos ou solventes.
- Não utilize o dispositivo se estiver danificado. Verifique se os cabos não estão danificados; os cabos deverão estar intactos.
- Se o cabo elétrico estiver danificado, deverá ser substituído pelo fabricante, pelo seu serviço pós-venda ou pessoal devidamente qualificado para evitar qualquer perigo.
- Não dobre, esmague ou estique excessivamente os fios ou os cabos.
- Os cabos podem provocar estrangulamento. Não coloque ou aperte os cabos à volta da cabeça ou do pescoço.
- Não ligue o dispositivo a qualquer outro equipamento ou a outra fonte de energia e verifique se a ficha elétrica não tem defeitos ou está danificada.
- Verifique se a voltagem indicada na ficha elétrica do dispositivo é a mesma voltagem da sua fonte de energia (se utilizar uma voltagem incorreta poderá provocar avarias que não são abrangidas pela garantia).
- Não utilize uma extensão.
- É proibido efetuar qualquer tipo de alterações no dispositivo. Não desmonte ou volte a montar o dispositivo.
VI. RECOMENDAÇÕES PARA A SUA UTILIZAÇÃO
Recomenda-se que utilize BLEPHASTEAM® duas vezes por dia ou conforme indicação do seu oftalmologista.
Deixe passar pelo menos quatro horas antes de repetir uma sessão de tratamento.
É importante que sejam seguidas as instruções de utilização (na secção VII) e as recomendações dadas pelo seu oftalmologista.
O seu dispositivo BLEPHASTEAM® destina-se a ser utilizado apenas por uma pessoa e deve ser limpo após cada utilização (ver secção VIII).
É compatível com uma voltagem entre 110 Volts e 220 Volts.
O dispositivo deve ser apenas utilizado com anéis BLEPHASTEAM® previamente humedecidos.
Estes anéis são de uso único e devem ser rejeitados após a sua utilização de forma a garantir a máxima eficácia do tratamento.
Os anéis BLEPHASTEAM® não são fornecidos com o dispositivo BLEPHASTEAM® e devem ser encomendados à THÉA PORTUGAL ou em www.blephasteam.com.
Entre cada sessão de tratamento, mantenha o seu dispositivo BLEPHASTEAM® dentro da bolsa que é fornecida.
Utilize este dispositivo à temperatura ambiente, entre 15°C e 30°C.
No local onde é guardado e durante o transporte, o dispositivo não deve ser sujeito a temperaturas inferiores a -10°C ou superiores a 40°C.
Para mais informações, por favor contacte o seu oftalmologista ou a THÉA PORTUGAL.
VII. INSTRUÇÕES PARA UTILIZAR O DISPOSITIVO BLEPHASTEAM®
Lave as suas mãos antes e depois de utilizar.
Utilize em olhos limpos sem qualquer maquilhagem.
As lentes de contacto ou os óculos devem ser retirados antes da sessão de tratamento.
Para uma maior eficácia e para que os anéis se mantenham corretamente colocados, não deve estar deitado quando utilizar o BLEPHASTEAM®.
Deverá estar sentado e não deverá mexer-se enquanto estiver a utilizar o dispositivo.
Pestaneje normalmente enquanto estiver a utilizar o dispositivo.
PASSOS 1 a 3: Pré-aquecimento
PASSOS 4 a 9: Tratamento
PASSOS 10 a 11: Limpeza/Conservação
Para efetuar uma sessão de tratamento siga todos os passos.
PRÉ-AQUECIMENTO:
PASSO 1
Ligue a ficha elétrica à tomada e deixe-a ligada ao longo de toda a sessão.
Deverá ouvir um sinal sonoro.
PASSO 2
Prima o botão On (botão azul do meio) com firmeza:
inicia-se o pré-aquecimento; acende-se a luz amarela.
NOTA: O botão On permite-lhe iniciar a fase de pré-aquecimento.
PASSO 3
Ao fim de 15 minutos, acende-se a luz verde:
terminou o pré-aquecimento.
O dispositivo encontra-se agora pronto para ser utilizado e pode iniciar a sessão de tratamento.
Dispõe de 45 minutos para iniciar a sua sessão.
Ao fim deste tempo, o dispositivo desliga-se e tem de ser reiniciado (recomece no passo 1).
TRATAMENTO:
PASSO 4
Humedeça dois anéis BLEPHASTEAM®, usando preferencialmente água mineral ou soro fisiológico (não ensope os anéis para evitar uma quantidade excessiva de água).
Mantenha-se longe da tomada durante este passo para que não entorne água em cima da ficha elétrica.
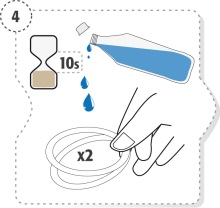
PASSO 5
Coloque estes anéis humedecidos dentro do dispositivo, um anel em cada lente.
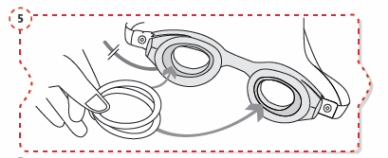
PASSO 6
Coloque o dispositivo sobre os seus olhos de forma confortável e ajuste-o em redor da sua cabeça usando a fivela para ajuste/fixação.
Os anéis devem manter-se bem colocados no interior das lentes dos óculos.
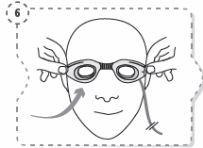
PASSO 7
Para iniciar a sessão, prima o botão On (botão azul do meio).
A luz verde deverá agora começar a piscar. A sessão de tratamento tem uma duração de 10 minutos.
Mantenha-se sentado durante a sessão.
Não desligue o dispositivo durante o tratamento.
NOTA: O botão On permite-lhe iniciar a sessão de tratamento.
PASSO 8
A sessão termina ao fim de 10 minutos.
O dispositivo pára automaticamente: ouvirá um sinal sonoro e a luz verde apaga-se.
PASSO 9
Retire o dispositivo, desligue a ficha da tomada e deite os anéis no lixo.
LIMPEZA / CONSERVAÇÃO:
PASSO 10
Massaje as suas pálpebras e, em seguida, limpe-as com uma compressa, como por exemplo, com BLEPHACLEAN®.
PASSO 11
Limpe o dispositivo com o pano azul fornecido ou com uma compressa embebida em álcool, tendo o cuidado de limpar todo o dispositivo e as lentes.
Guarde o dispositivo e o pano azul dentro da bolsa.
Caso a sessão seja interrompida por qualquer motivo, desligue o dispositivo e recomece a partir do Passo 1.
VIII. LIMPAR E GUARDAR O SEU DISPOSITIVO BLEPHASTEAM®
Quando limpar o dispositivo, este deverá estar desligado da tomada.
Após cada utilização, deve limpar o dispositivo com o pano azul fornecido ou com uma compressa embebida em álcool.
O dispositivo deve depois ser guardado na sua bolsa.
IX. GARANTIA E SERVIÇOS
A garantia é válida durante UM ANO desde a data da sua aquisição.
A garantia cobre qualquer falha, dano do material ou defeito de fabrico desde que o dispositivo tenha sido usado em conformidade com as instruções descritas neste folheto.
Esta garantia não exclui a aplicação das atuais garantias legais em função da legislação nacional associada à venda de produtos de consumo.
A garantia não é aplicável nos seguintes casos:
- Danos durante o transporte,
- Utilização incorreta ou negligente,
- Manuseamento incorreto,
- Modificação do sistema,
- Manutenção inadequada,
- Utilização de uma voltagem errada,
- Relâmpagos,
- Infiltração de areia ou água.
X. MANUTENÇÃO E INSTRUÇÕES EM CASO DE AVARIA
Inspeção preventiva
Verifique se o seu dispositivo BLEPHASTEAM® está a funcionar corretamente, pelo menos, uma vez por ano ou se o dispositivo não for usado durante um longo período de tempo (mais de um mês).
Para tal, siga os seguintes passos:
- Efetue os passos 1 a 3 da secção VII. INSTRUÇÕES DE UTILIZAÇÃO.
- Tempo de pré-aquecimento (enquanto a luz amarela estiver acesa).
- Registe o tempo de pré-aquecimento na tabela abaixo, na coluna referente ao “Tempo de pré-aquecimento”. O tempo registado deve estar compreendido entre 14 e 16 minutos.
- Verifique se os óculos produzem calor.
- Efetue o passo 7 da secção VII. INSTRUÇÕES DE UTILIZAÇÃO, para iniciar uma sessão de 10 minutos.
- Tempo de duração da sessão de tratamento (enquanto a luz verde estiver a piscar).
- Registe o tempo de duração da sessão de tratamento na tabela abaixo, na coluna referente ao “Tempo de aquecimento”. O tempo registado deve estar compreendido entre 9 e 11 minutos.
Se um dos 2 tempos registados não estiver dentro dos limites indicados OU se o dispositivo não produzir qualquer calor, então não se encontra operacional.
Neste caso, não utilize o dispositivo e devolva-o à THÉA PORTUGAL na sua embalagem original, incluindo este folheto, a cópia da fatura de compra e o motivo para a devolução do dispositivo.
| Data |
Tempo de pré-aquecimento (Luz amarela) 14 min < duração < 16 min |
Tempo de aquecimento (Luz verde a piscar) 9 min < duração < 11 min |
O dispositivo não requer qualquer manutenção ou calibração.
O dispositivo tem 7 anos de vida útil.
Em caso de falha ou mau funcionamento, por favor devolva-o à THÉA PORTUGAL na sua embalagem original e inclua uma cópia da fatura de compra e o motivo para a devolução do dispositivo.
Para qualquer outro problema ou questão acerca do funcionamento do equipamento, por favor contacte a THÉA PORTUGAL.
XI. FABRICANTE E DISTRIBUIDORES NA EUROPA
Laboratoires THÉA
12, rue Louis Blériot
63017 Clermont-Ferrand
CEDEX 2 - FRANÇA
DISTRIBUIDOR:
Ver lista no final do folheto
XII. INFORMAÇÕES TÉCNICAS
O dispositivo BLEPHASTEAM® funciona com uma voltagem entre 110 Volts e 220 Volts.
Referência do dispositivo BLEPHASTEAM®: T2420. Fornecido com uma bolsa e um pano de limpeza azul.
Os anéis do BLEPHASTEAM® não são fornecidos com o BLEPHASTEAM® e têm de ser encomendados à THÉA PORTUGAL ou em www.blephasteam.com.
Caixa de controlo/cabo elétrico:
Ficha elétrica
Referência: FW7333SM/09 (artigo número: 1831144).
A voltagem mínima é de 9V e a intensidade da corrente é de 0,8A.
Eliminação deste dispositivo elétrico
Devem seguir-se as disposições legais em vigor (Diretiva 2002/96/CE) na eliminação deste dispositivo.
Deverá ser colocado num ponto de reciclagem ou poderá ser devolvido à THÉA PORTUGAL caso seja adquirido um novo dispositivo.
Símbolos usados e sua definição
| Parte aplicada: Tipo BF (nível de segurança especificado pelo padrão IEC 60601-1) | |
| Dispositivo de Classe II | |
 |
Dispositivo sensível à humidade |
| Consulte o folheto de instruções | |
 |
Data de fabrico |
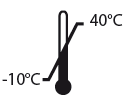 |
Limites de temperatura aos quais o dispositivo pode ser exposto com segurança |
| Número do lote | |
| Referência do produto | |
 |
Data de validade |
 |
Não deite fora no lixo doméstico |
 |
Fabricante |
Electromagnetic interference
| Conducted Emissions |
EN60601-1-2 group 1 Class B, CISPR 11 group 1 Class B |
| Radiated Emissions |
EN60601-1-2 Class B, CISPR 11 group 1, Class B |
| Line Frequency Harmonics | Rated power less than 75 W |
| Voltage fluctuation/Flicker | EN61000-3-3 |
| Static Discharge Immunity |
EN61000-4-2, 6kV contact discharge, 8 kV air discharge |
| Radiated RF Immunity | EN61000-4-3, 3V/m. |
| EFT/Burst Immunity | EN61000-4-4, 2kV/5kHz. |
| Line Surge Immunity |
EN61000-4-5, 1kV differential, 2kV common-mode |
| Conducted RF Immunity | EN61000-4-6, 3V, 130dB (µV) |
| Power Frequency Magnetic Field Immunity | EN61000-4-8, 10A/m |
| Voltage Dip Immunity | EN61000-4-11, Criteria B |
Data de atualização do folheto de instruções: Junho 2013

Pálpebras
Blephagel
BLEPHAGEL® 30g gel
Gel suave para limpeza diária das pálpebras.
Adequado para pele sensível, irritada e com tendência atópica.
Testado sob controlo oftalmológico.
Sem conservantes, sem perfume, sem álcool**
1. O QUE É BLEPHAGEL E QUAIS OS SEUS BENEFÍCIOS?
BLEPHAGEL® é um gel hipoalergénico** formulado para a limpeza suave diária da pele sensível, irritada e com tendência atópica.
BLEPHAGEL® é especificamente formulado para:
- Refrescar, suavizar e acalmar as pálpebras.
- Remover sujidade, lágrimas, descamações secas, pólen e maquilhagem.
- Respeitar a hidratação natural da pálpebra.
- Não deixar resíduos e não ser gorduroso ou pegajoso.
BLEPHAGEL foi testado sob controlo oftalmológico e demonstrou uma excelente tolerância.
BLEPHAGEL faz parte da gama BLEPHA da Théa, uma gama 100% dedicada ao cuidado das pálpebras para o bem-estar das suas pálpebras e manutenção de uma boa saúde visual
AQUA, POLOXAMER 188, PEG-90, BHT, CARBOMER, SODIUM HYDROXIDE.
2. O QUE POSSIBILITA O BLEPHAGEL NÃO TER CONSERVANTES?
A Théa, especializada em produtos oftalmológicos, foi a pioneira e continua a ser uma das líderes mundiais em produtos oftalmológicos sem conservantes.
BLEPHAGEL® beneficia de:
1. Uma formulação específica apenas com os ingredientes essenciais, cuidadosamente selecionados de acordo com a sua eficácia, para garantir uma melhor tolerância e segurança (sem conservantes, pseudo-conservantes ou irritantes).
2. Da tecnologia da bisnaga ser feita de um doseador airless que fornece uma dose precisa e regular do gel e permite uma aplicação simples e fácil.
3. Um processo de fabrico com elevado nível que garante a qualidade do gel.
3. O QUE É QUE OS OFTALMOLOGISTAS RECOMENDAM FAZER NOS DISTÚRBIOS NAS PÁLPEBRAS?
Os oftalmologistas recomendam a realização de uma higiene diária das pálpebras para ajudar nas seguintes situações:
- Blefarites (anteriores e posteriores): inflamação das pálpebras,
- Olho seco associado à disfunção das glândulas de meibomius (glândulas localizadas no interior das pálpebras que não funcionam corretamente),
- Conjuntivite alérgica: resposta inflamatória a alergenos,
- Lesões no olho e nas pálpebras devidas a:
- Eczema (dermatite atópica): doença da pele da face que causa comichão, pele seca, erupções cutâneas, manchas descamativas,
- Acne rosácea: doença da pele no rosto que causa vermelhidão,
A higiene diária das pálpebras também pode ser recomendada antes da cirurgia apenas se aconselhada pelo seu médico/oftalmologista, quando é necessária uma limpeza cuidadosa e completa das pálpebras e da base das pestanas.
4. QUEM PODE UTILIZAR BLEPHAGEL?
BLEPHAGEL é adequado para pele sensível, irritada e com tendência atópica.
BLEPHAGEL pode ser recomendado para olhos sensíveis e utilizadores de lentes de contacto.

BLEPHAGEL não é gorduroso nem pegajoso e não necessita de ser enxaguado após a utilização.
BLEPHAGEL contém aproximadamente 60 aplicações.
Não aplicar diretamente nos olhos.
6. COMO CONSERVAR BLEPHAGEL?
Manter fora da vista e do alcance das crianças.
Rejeitar a bisnaga 4 meses após a primeira abertura.
Não conservar acima de 25°C.
Não cortar a bisnaga (risco de contaminação por bactérias).
Made in Europe
©2020, Laboratoires Théa. All rights reserved.
Théa®, BLEPHAGEL® são marcas registadas dos Laboratoires Théa.
*Sem etanol
**O produto foi desenvolvido para reduzir a reação na pele.

Pálpebras
Blephasol

Blephasol
Loção micelar.
Higiene diária das pálpebras sensíveis.
Frasco de 100 ml.
Apresentação:
Loção micelar para limpeza das pálpebras sensíveis.
Frasco de 100 ml.
Para aplicação cutânea.
Propriedades e indicações:
BLEPHASOL é recomendado para a higiene diária das pálpebras sensíveis e, nomeadamente, do bordo palpebral livre.
Graças às suas propriedades micelares, BLEPHASOL actua mecanicamente, eliminando as impurezas, crostas e maquilhagem das pálpebras e da raiz das pestanas.
Não contém álcool (etanol).
BLEPHASOL limpa suavemente, sem irritar os olhos nem agredir as camadas exteriores da epiderme, que asseguram a protecção da pele contra as agressões exteriores (frio, sol, suor, alergénios, poluentes, etc.).
Não perfumado e não gorduroso. Não é necessário lavar com água após utilização.
BLEPHASOL pode ser recomendado aos utilizadores de lentes de contacto.
Modo de emprego:
Em média, duas vezes por dia, de manhã e à noite, ou sempre que deseje limpar as pálpebras.
Para limpeza e desmaquilhagem das pálpebras sensíveis:
Em frente de um espelho:
| 1- Aplicar delicadamente uma compressa hidrófila humedecida com BLEPHASOL nas pálpebras e na raiz das pestanas, com o olho fechado. | |
| 2- Massajar delicadamente as pálpebras com pequenos movimentos circulares, a fim de eliminar todas as impurezas e extrair o conteúdo das glândulas palpebrais. | |
| 3- Puxar ligeiramente as pálpebras e, em seguida, aplicar delicadamente no bordo interno das mesmas, com uma nova compressa ou um cotonete humedecidos com BLEPHASOL para eliminar as impurezas alojadas na raiz das pestanas. | |
| 4- Repetir estas etapas em relação ao outro olho, utilizando uma nova compressa. |
Não é necessário lavar com água após utilização.
Precauções especiais:
Não ingerir, não aplicar diretamente nos olhos, manter afastado das crianças.

Pálpebras
Blephaclean

Blephaclean
Indicações de utilização:
HIGIENE DAS PÁLPEBRAS BLEPHACLEAN
Destina-se à higiene diária das pálpebras, especialmente:
- Se tem uma pele, pálpebras ou olhos sensíveis,
- Se necessita de remover poeiras, pólen ou secreções das suas pálpebras. Tal poderá fazer parte dos cuidados a ter quando tem uma alergia ou infeção nos seus olhos,
- Se tem uma inflamação ou infeção nas pálpebras tal como blefarite, disfunção das glândulas de Meibomius, terçolho ou chalázio. Estas condições podem causar sintomas de secura ocular tais como, comichão, sensação de areia, dor e/ou vermelhidão nos olhos,
- Quando necessita de limpar as pálpebras antes de procedimentos oftalmológicos, tais como injeções terapêuticas nos seus olhos (injeções intravítreas), cirurgia laser nos olhos (LASIK ou PRK) ou cirurgia da catarata. Como é bem tolerado, BLEPHACLEAN pode também ser utilizado após a cirurgia, tendo em consideração as indicações do seu médico ou oftalmologista.
BLEPHACLEAN mantém as suas pálpebras limpas, respeitando ao mesmo tempo a pele em redor dos seus olhos e as suas bactérias naturais.
Antes de uma cirurgia ocular, utilize duas vezes por dia durante 5 dias ou como indicado pelo seu médico ou farmacêutico.
Para as outras situações, utilize duas vezes por dia de manhã e à noite durante 3 semanas e depois uma vez por dia durante 3 meses ou como indicado pelo seu médico ou farmacêutico.
Algumas situações a nível das pálpebras são crónicas e poderá ter de manter a higiene das pálpebras durante um longo período de tempo.
Não é necessário enxaguar após a utilização.
BLEPHACLEAN pode ser utilizado em crianças a partir dos 3 meses de idade.
O produto foi testado sob controlo oftalmológico.
Se utiliza lentes de contacto:
retire as lentes antes de utilizar BLEPHACLEAN.
Instruções de utilização:
1. Higiene das pálpebras e pestanas:
Abra a saqueta e desdobre o toalhete que se encontra no seu interior.
Com o olho fechado, aplique delicadamente o toalhete nas pálpebras e na base das pestanas.
Massaje cuidadosamente as pálpebras com pequenos movimentos circulares, removendo com suavidade quaisquer crostas, secreções ou maquilhagem.
2. Higiene dos bordos das pálpebras:
Em frente a um espelho, abra cuidadosamente a pálpebra, puxando ligeiramente as pestanas.
Esfregue delicadamente o bordo palpebral com um toalhete enrolado.

Repita os passos acima no outro olho utilizando um toalhete novo.
Precauções de utilização e advertências:
Apenas para aplicação na pele, não aplique diretamente nos olhos. Não utilize se tem alergia a qualquer um dos componentes.
BLEPHACLEAN é um produto estéril, para uma utilização única.
Não utilize novamente, pois tal pode fazer com que os toalhetes atuem menos bem e pode aumentar o risco de infeção.
Não utilize o toalhete se a saqueta estiver aberta ou danificada.
A cor do toalhete (loção) pode variar.
Tal é uma situação normal e deve-se aos extratos naturais da planta.
Se os seus sintomas se agravarem deixe de utilizar BLEPHACLEAN e contacte o seu médico ou farmacêutico.
Não utilize após o prazo de validade indicado na embalagem exterior e na saqueta.
O prazo de validade referese ao produto na sua embalagem intacta e conservado corretamente.
Os toalhetes devem ser rejeitados no lixo doméstico após a utilização.
Conservar entre 8 e 25°C.
MANTER FORA DA VISTA E DO ALCANCE DAS CRIANÇAS.
Possíveis efeitos adversos:
Foram notificados os seguintes eventos com BLEPHACLEAN:
- sensação de queimadura ou comichão durante alguns minutos após a utilização, vermelhidão ou inchaço das pálpebras, reações alérgicas.
Descrição do produto:
Toalhete estéril, impregnado com uma loção.
A loção contém:
Capriloil glicina, Hialuronato de sódio, PEG 8, Poloxamero 184, Polissorbato 20, Propilenoglicol, extrato de hidrocotil (propilenoglicol, extrato de Centelha asiática e água), Complexo-seboregulador sem conservantes (propilenoglicol, extrato de raiz de Iris florentina, sulfato de zinco, PPG-5 Ceteth 20 e palmitato de retinol (Vitamina A), álcool e água), PEG 6 glicerídeos caprílicos/ cápricos (i.e. Macrogol 6 glicerol caprilocaprato), Fosfato dipotássico, Hidróxido de sódio, Dihidrogenofosfato de potássio, Água purificada.
Data de revisão das instruções de utilização:
Fevereiro 2016
Distribuidor: Théa Portugal, S.A.
Rua Pedro Álvares Cabral, 24 – 5ºF,
2670-391 Loures,
Portugal
Fabricante
Laboratoires THEA
12, rue Louis Blériot 63017
Clermont-Ferrand Cedex 2
França

Pálpebras
Blephademodex

Indicações de utilização:
BLEPHADEMODEX é recomendado:
- para a higiene diária das pálpebras em caso de infeção por Demodex,
- para o alívio dos sintomas de infeção ou inflamação da pálpebra causada por Demodex,
O demodex é o parasita microscópico da pele mais comum. A infeção por Demodex contribui para a inflamação da pálpebra (blefarite). O Demodex não é removido pelos agentes de limpeza diários normalmente utilizados. A presença de películas cilíndricas, que permanecem na base das pestanas, é um importante indicador de infeção por Demodex.
BLEPHADEMODEX remove crostas, películas, impurezas e agentes infeciosos das pálpebras e pestanas. BLEPHADEMODEX melhora os sintomas associados à infeção por Demodex, tais como a comichão, a sensação de ardor, a secura e a inflamação dos bordos das pálpebras, através da remoção dos parasitas Demodex. Há uma diminuição da vermelhidão dos bordos das pálpebras e da sensação de corpo estranho.
A pele sensível torna-se mais suave e revitalizada por ação do hialuronato de sódio que é um agente hidratante natural da pele.
BLEPHADEMODEX está formulado com Terpeno-4-ol e é bem tolerado pois é isento de detergentes e conservantes.
BLEPHADEMODEX tem uma fórmula sem perfume, no entanto, devido aos seus componentes pode sentir um cheiro fresco, que se dissipa ao fim de poucos minutos.
A cor do toalhete (loção) pode variar. Tal é normal e deve-se aos extratos naturais da planta.
Dose:
Sintomas ligeiros a moderados: utilizar uma vez ao dia, preferencialmente à noite, durante 4 semanas.
Sintomas moderados a graves: utilizar duas vezes ao dia, de manhã e à noite, durante 4 semanas.
O seu profissional de saúde poderá adaptar a posologia e a duração do tratamento de acordo com a gravidade dos sintomas.
Tal como com outros tratamentos semelhantes para a blefarite, o seu profissional de saúde poderá recomendar a utilização concomitante de lágrimas artificiais.
BLEPHADEMODEX pode ser utilizado a partir dos 18 anos de idade.
Instruções de utilização:
Se utiliza lentes de contacto: tem de retirar as lentes antes de utilizar BLEPHADEMODEX.
Abra a saqueta e desdobre o toalhete que se encontra no seu interior.
Coloque o toalhete na ponta do dedo indicador e enrole-o à volta do dedo.
Com o olho fechado, aplique delicadamente o toalhete nas pálpebras e na base das pestanas.
Massaje cuidadosamente as pálpebras com pequenos movimentos circulares, removendo com suavidade quaisquer crostas, películas ou secreções.
Repita no outro olho utilizando um toalhete novo.
BLEPHADEMODEX não é oleoso e não é necessário enxaguar após a utilização.
Mantenha o seu olho fechado durante 15 segundos após a aplicação.
Precauções de utilização e advertências:
Apenas para aplicação na pele.
Evite tocar com o BLEPHADEMODEX no olho pois poderá sentir uma sensação de ardor.
Se tal acontecer, enxague com água estéril ou soro fisiológico.
BLEPHADEMODEX é um produto estéril, para uma utilização única. Não utilize novamente, pois tal pode originar um reduzido efeito de limpeza e aumentar risco de infeção.
Não utilize o toalhete se a saqueta estiver aberta ou danificada.
Não utilize após o prazo de validade indicado na embalagem exterior e na saqueta. O prazo de validade refere-se ao produto na sua embalagem intacta e conservado corretamente.
Os toalhetes utilizados devem ser eliminados no lixo doméstico.
Deixe de utilizar BLEPHADEMODEX e contacte o seu profissional de saúde se os seus sintomas se agravarem ou se ocorrer alguma reação alérgica.
Contraindicações:
Não utilize se tiver menos de 18 anos de idade pois não existem dados disponíveis.
Não utilize se estiver grávida ou a amamentar pois não existem dados disponíveis.
Não utilize se tiver alergia a qualquer um dos componentes.
Conservar entre 8 e 25°C.
MANTER FORA DA VISTA E DO ALCANCE DAS CRIANÇAS.
Possíveis efeitos secundários:
Foram notificados os seguintes efeitos secundários: sensação de ardor após a utilização.
Se tiver quaisquer efeitos secundários, fale com o seu médico, oftalmologista, farmacêutico ou com o fabricante.
Descrição do produto:
Toalhete estéril, impregnado com uma loção.
A loção contém: PEG-8, Polysorbate 80, Poloxamer 184, Terpinen-4-ol, PEG-6 caprylic/capric glycerides Macrogol 6 glycerol caprylocaprate, Sodium hyaluronate, Dipotassium phosphate, Potassium dihydrogen phosphate, Purified water.
Data de revisão das instruções de utilização:
Dezembro 2017
Fabricante
Laboratoires THEA
12, rue Louis Blériot
63017 Clermont-Ferrand Cedex 2
França

Pálpebras
BLEPHADERM®

BLEPHADERM®
CUIDADO DAS PÁLPEBRAS
1. O QUE É BLEPHADERM® E QUAIS OS SEUS BENEFÍCIOS?
BLEPHADERM 5 em 1creme para pálpebras e contorno de olhos:
1. Suaviza
2. Hidrata
3. Repara
4. Protege
5. Reduz o inchaço
BLEPHADERM® é um creme para suavizar a pálpebra e o contorno do olho que proporciona um efeito suavizante imediato e uma
hidratação prolongada. Reduz irritações (vermelhidão e picadas), sensações de prurido e inchaço. BLEPHADERM é o primeiro produto da Théa para cuidar do olho, sem conservantes, sem perfume e estéril, indicado para a pele sensível, seca, irritada e com tendência atópica.
BLEPHADERM® proporciona conforto imediato e prolongado. Reforça a barreira da pele e as defesas naturais para uma melhor proteção contra fatores externos.
O aspeto da pele é melhorado: o contorno do olho fica uniforme e reduz o aparecimento e o tamanho dos papos sob os olhos.
EFICÁCIA APROVADA PELO CONSUMIDOR1
Os utilizadores notaram: uma redução da vermelhidão, da irritação e da sensação de prurido logo após a aplicação em 100% dos utilizadores. A pele fica imediatamente suavizada, hidratada, protegida e mais confortável (100%). Desde o primeiro dia de utilização, a pele fica menos sensível para 100% e reparada para 91% dos utilizadores. Este cuidado protege a pele contra fatores externos (95%).
Ingredientes
AQUA (WATER), OLEYL ERUCATE, TREHALOSE, SODIUM ACRYLATES COPOLYMER, LECITHIN, OPHIOPOGON JAPONICUS ROOT EXTRACT, HYDROLYZED HYALURONIC ACID, MALTODEXTRIN, TOCOPHEROL, SODIUM HYDROXIDE, PHYTIC ACID, GLYCINE SOJA (SOYBEAN) OIL, HELIANTHUS ANNUUS SEED OIL.
2. COMO SÃO INOVADORES A FÓRMULA E O DISPOSITIVO DE BLEPHADERM?
BLEPHADERM®, um creme inovador e patenteado para cuidar das pálpebras e do contorno de olhos, foi formulado e testado por
oftalmologistas e dermatologistas.
BLEPHADERM® beneficia da sinergia de um trio de ingredientes inovadores de origem natural:
• O ácido hialurónico, pelas suas propriedades regeneradoras e capacidade de retenção de água proporciona uma hidratação
profunda e contínua dos tecidos.
• A trealose bio-protetora repara e protege a epiderme.
• O extrato da raiz de Ophiopogon japonicus reforça a barreira da pele, respeita a microbiota e mantém a hidratação.
A formulação de BLEPHADERM beneficia da tecnologia Sterile CosmeticsTM que permite obter um creme estéril e sem conservantes.
A inovação Sterile CosmeticsTM combinada com:
• Uma seleção rigorosa e limitada de apenas ingredientes essenciais para obtenção de formulações seguras e eficazes
• Um processo único em dermocosmética: esterilização ao longo do processo de fabrico (com uma esterilização completa do
equipamento de produção, um método patenteado de esterilização de formulações, acondicionamento em ambiente estéril)
• Um inovador sistema de selagem: o sistema D.E.F.I (Device for Exclusive Formula Integrity) é uma abordagem revolucionária no
design do acondicionamento em dermocosmética, desenvolvido e patenteado pela Pierre Fabre. O sistema perfeitamente estanque ajuda a preservar uma formulação completamente estéril, impedindo que bactérias e todos os outros microrganismos contaminem a formulação durante toda a utilização do produto. *Patente submetida
3. QUEM PODE USAR BLEPHADERM®?
BLEPHADERM é adequado para todos os tipos de pele e particularmente para pele sensível, seca, irritada e com tendência atópica.
BLEPHADERM é adequado para olhos sensíveis e para utilizadores de lentes de contacto.
BLEPHADERM foi testado sob controlo dermatológico e oftalmológico, tendo demonstrado uma excelente tolerância.
4. COMO USAR BLEPHADERM®?
Antes de usar BLEPHADERM, limpe a sua pele com um produto de limpeza das pálpebras, de preferência da gama BLEPHA da Théa.

BLEPHADERM® pode ser aplicado diariamente em toda a face. Tem uma textura agradável na pele, não é pegajoso e não é oleoso. É uma excelente base para maquilhagem.
BLEPHADERM® contém aproximadamente 200 aplicações.
Não aplique diretamente nos olhos.
5. COMO CONSERVAR BLEPHADERM®?
Manter fora da vista e do alcance das crianças.
Rejeitar a bisnaga 2 meses após a primeira abertura.
Não conservar acima de 30ºC.
Não refrigerar, não congelar.
Não corte a bisnaga (risco de contaminação bacteriana).
Fabricado em França
©2023, Laboratoires Théa. Todos os direitos reservados.
Théa®, BLEPHADERM® são marcas registadas dos Laboratoires THEA.